ABSTRACT
This article aims to review the physiopathology, diagnosis and treatment of cystic fibrosis-related dyslipidemia (CFD). Bibliographic searches of the Medline and Latin American and Caribbean Health Sciences Literature databases were made (year range, 1987-2007), and the most representative papers on the theme were selected. The characteristic symptoms of CFD are hypertriglyceridemia-with or without hypocholesterolemia-and essential fatty acid deficiency. The principal CFD risk factors are pancreatic insufficiency, high-carbohydrate diet, liver diseases, inflammatory state and corticosteroid therapy. There are no specific recommendations regarding screening, which is typically performed based on the diagnosis, and at regular intervals, and more frequently in individuals belonging to high-risk groups. Treatment includes a balanced diet, micronutrient supplementation, and regular physical exercise according to individual tolerance. In the great majority of the cases, CFD-related hypertriglyceridemia does not reach values for which the use of hypolipidemic drugs is indicated. We conclude that there are few articles in the literature regarding the frequency, etiology and management of CFD. Preventive and therapeutic recommendations for hypertriglyceridemia are extrapolated from studies in individuals without cystic fibrosis. Further research is necessary to investigate the association of essential fatty acid deficiency and the physiopathology of cystic fibrosis. Since hypertriglyceridemia is an important risk factor for coronary artery disease, prospective studies will contribute for a better understanding of the natural history of this condition and define how to prevent and treat it.
Keywords:
Cystic fibrosis; Dyslipidemias; Hypertriglyceridemia; Fatty Acids, nonesterified.
RESUMO
Esse artigo tem por objetivo rever o conhecimento atual sobre a fisiopatologia, o diagnóstico e a abordagem da dislipidemia relacionada à
fibrose cística (DFC). A pesquisa bibliográfica utilizou os bancos de dados Medline e Literatura Latino-Americana e do Caribe em Ciências
da Saúde (1987-2007), selecionando os artigos mais relevantes sobre o tema. A DFC é caracterizada por hipertrigliceridemia e/ou hipocolesterolemia
e deficiência de ácidos graxos essenciais. Seus principais fatores de risco são: insuficiência pancreática, dieta rica em carboidratos,
hepatopatias, estado inflamatório e corticoterapia. Não existem recomendações específicas sobre a triagem, que habitualmente é realizada a
partir do diagnóstico e, em intervalos regulares, com maior freqüência, nos indivíduos pertencentes aos grupos de risco. O tratamento inclui:
dieta balanceada, reposição de micronutrientes, vitaminas e fibras, além de exercício físico regular de acordo com a tolerância individual. Na
grande maioria dos casos, a hipertrigliceridemia da DFC não atinge valores que indiquem o uso de hipolipemiantes. Conclui-se que existem
poucos trabalhos na literatura sobre a freqüência, etiologia e manejo da DFC. A recomendações preventivas e terapêuticas para a hipertrigliceridemia
são extrapoladas de diretrizes para indivíduos sem fibrose cística. Mais pesquisas são necessárias para investigar a associação
da deficiência de ácidos graxos essenciais com a fisiopatologia da fibrose cística. Como a hipertrigliceridemia é um importante fator de risco
para doença arterial coronariana, estudos prospectivos irão contribuir para o melhor entendimento da história natural dessa complicação
bem como definir maneiras de preveni-la e tratá-la.
Palavras-chave:
Fibrose cística; Dislipidemias; Hipertrigliceridemia; Ácidos graxos não-esterificados.
IntroductionCystic fibrosis (FC) is a multisystemic hereditary disorder primarily characterized by exocrine pancreatic insufficiency, obstruction and infection of the airways.(1) Clinical manifestations can be explained, for the most part, by the chronic inflammation and by the dysfunction of the cystic fibrosis transmembrane regulator protein (CFTR), with malfunctioning of the chloride channel.(2) Among the many mutations described in the CF gene, the most frequently found is ΔF508.(3)
As a result of the increases in life expectancy due to the advancements in treatment, some complications unobserved before have begun to be diagnosed, such as diabetes mellitus, liver disease, osteoporosis and, more recently, dyslipidemia.(4) Some individuals with CF present a dyslipidemic pattern characterized by hypertriglyceridemia with or without hypocholesterolemia.(5) The most frequently observed alteration is hypertriglyceridemia, although these patients have an increase in energy expenditure, poor intestinal absorption of fat and reduction in food intake.(6)
Although hypertriglyceridemia is one of the risk factors for atherosclerotic cardiovascular disease, little attention has been given to the impact of this complication in the morbidity of patients with CF. This fact becomes more relevant when there is an association with CF-related diabetes mellitus, since hyperglycemia contributes for the atherogenesis through glycation and peroxidation of the low-density lipoprotein (LDL).(4,7)
Due to the lack of studies on the theme, the objective of the present article is to conduct a critical review on the current knowledge of physiopathology, diagnosis and approach of dyslipidemia in patients with CF. In the present study, this complication was named cystic fibrosis-related dyslipidemia (CFD).
Medline and the Latin American and Caribbean Health Sciences Literature databases were used in the bibliographic search, selecting the most relevant articles on the theme published in the period of 1987 to 2007.
Normal lipid metabolismCholesterol and triglycerides (TG; or triacylglycerol) are the principal plasma lipids. Cholesterol is fundamental for the synthesis of steroid hormones, synthesis of bile acids and formation of cell membranes. The TGs are the principal energy reserve of the body. Essential fatty acids (for instance, linoleic and linolenic), participate in the development and functioning of the central nervous system, immune system and vascular function. However, since these are inadequately produced by the mammal cells, maintenance of their serum levels depends on adequate intake and absorption.
Since they are practically insoluble in water, cholesterol and TG are transported in plasma associated with proteins, forming soluble complexes named lipoproteins.(8) Lipoproteins have spherical form and are formed by the nucleus and by the external layer. The nucleus contains principally cholesterol and TG esters.(8) The external layer is composed of phospholipids and free cholesterol.(9) On the surface, there are apoproteins or apolipoproteins, which bind to specific receptors of the cell responsible for the metabolism of lipoproteins.(8,10)
Lipoproteins are classified according to their electrophoretic mobility and density (the greater the quantity of apoproteins, the greater the density, and the greater the quantity of TG, the lower the density).(9) The principal lipoproteins are the following: chylomicrons (CM), very low-density lipoprotein (VLDL), intermediate density lipoprotein, low-density lipoprotein (LDL) and high-density lipoprotein (HDL). Table 1 shows the properties of the principal lipoproteins.

In the intestine, the cholesterol and TG of the diet are emulsified by bile acids and hydrolyzed by pancreatic lipase. TGs are broken into fatty acids and monoglycerides, and the esters of cholesterol in fatty acids and non-esterified cholesterol. In the intestinal cell, the monoglycerides are re-esterified in TG and stored in the CMs. The ApoC-II, ApoB-48 and Apo-E are the principal apolipoproteins of the CMs. The CMs are secreted in the lymph through the thoracic duct and taken to the systemic circulation through the vena cava. In the capillary endothelium, the CMs bind to the lipase lipoprotein through the ApoC-II, releasing the TGs as monoglycerides and free fatty acids.(8) The muscle uses the free fatty acids and monoglycerides to produce energy. The adipose tissue uses those to re-synthesize TG and store them in the adipose cells.(9) The CM resulting of this metabolism, CM remnant, smaller and richer in cholesterol, is captured by the liver. Normally, the CMs are only found in the plasma in the postprandial period, for they are the transporters of the TGs of the diet.(8) In the liver, there is the synthesis of the TG and the cholesterol, which are incorporated to the VLDLs, and excreted afterwards.(10) The VLDL transports TG from the liver to the peripheral tissues. In the cell membrane, they bind to the lipoprotein lipase through the ApoC-II in order to liberate the TGs, forming the intermediate density lipoproteins.(8) The intermediate density lipoproteins are removed by the liver via ApoE where they are transformed in LDL. The LDL is the main transporter of cholesterol to the cells.(8) In the extrahepatic tissues, including the subendothelial space where the protecting effect of the circulating antioxidants (for instance, vitamin E) is lost, the LDL is oxidized, initiating the formation of the atheromatous plaques. The HDL is produced in the liver and intestine and acts in the reverse transport of cholesterol, the process in which the HDL captures the excess of cholesterol and TG liberated to the cells and transports them to the liver.(11)
Physiopathology of cystic-fibrosis related dyslipidemiaThe physiopathology of CFD is not totally elucidated yet. The most studied mechanisms are the following: carbohydrate-rich and low-fat diet, fat-rich diet, liver disease, elevation of the pro-inflammatory cytokines and use of corticosteroids, probably acting in combination.(5)
Previous studies demonstrate no association of the dyslipidemia with intolerance to glucose, diabetes mellitus, thyroid disease, specific genotypes of CF, pulmonary function, concentration of the C-reactive protein, age, gender, weight, body mass index and arterial pressure.(5,12)
Carbohydrate-rich, low-fat dietPatients with exogenous pancreatic insufficiency and inadequate enzymatic replacement do not tolerate diets with normal fat level due to poor intestinal absorption, and, as a defense mechanism, consume an excessive amount of carbohydrates.(5,13) The consumption of these low-fat carbohydrate-rich diets can be one of the causes of the hypertriglyceridemia related to CF. In these cases, the excessive and chronic consumption of carbohydrates makes the liver exceed its capacity of synthesis and storage of glycogen, deviating the metabolic access to the production of TG.(14) Other explanations would be the decrease of the activity of the lipoprotein lipase and the increase of the intestinal absorption of glucose.(15)
Fat-rich dietIn order to study the repercussion of a fat-rich diet on the serum lipids, a study compared the lipid profile of adults with CF, with and without exogenous pancreatic insufficiency, with healthy controls.(16) It was observed that patients with CF and pancreatic insufficiency, despite consuming great quantities of fat and cholesterol, presented no worsening in the lipid profile, in contrast to those without exogenous pancreatic insufficiency, in which the same atherogenic risk of the general population was observed. That is, CF patients with normal exogenous pancreatic function present elevated risk of atherogenesis, similar to the general population, when consuming fat-rich diets. However, this hypothesis has been controversial since a study in which no significant difference in the lipid profile of patients with CF was observed when compared to the control group, despite the use of a fat-rich diet,(17) and in another study it was reported that, even with enzymatic supplementation, the absorption of the fat of the diet varied 79-93%.(18)
Hepatic dysfunctionLiver disease is considered a serious comorbidity of the CF, altering its prognostic and the life quality.(19) The prevalence depends on the criteria used: from 1.4-7% by clinical evaluation, to more than 20% if biochemical and ultrasonographic methods are used.(19) Liver disease results from the hepatic steatosis, which is usually attributed to the accumulation of TG and LDL in the hepatic parenchyma due to the reduction in the synthesis of apolipoproteins and, in some cases, due to carnitine deficiency.(20) Depending on its severity, the liver disease can contribute to the hypertriglyceridemia.(21)
Increase in the serum levels of the tumor necrosis factor-alpha and of C-reactive proteinPro-inflammatory cytokines, such as the tumor necrosis factor-alpha, are mediators of hyperlipidemia in situations of infection or stress by inhibiting the activity of the lipoprotein lipase (decreasing the clearance of TG) and stimulating the hepatic lipogenesis.(22) Therefore, the hypertriglyceridemia of the CF can be related to the chronic inflammatory status exacerbated by frequent outbreaks of infection. C-reactive protein, another inflammatory mediator, did not associated with dyslipidemia.(5) Recent studies have drawn attention to the possibility of treatments with systemic anti-inflammatory drugs being beneficial to patients with chronic inflammatory diseases, such as CF, improving the nutritional status and reducing insulin resistance, one of the factors which contribute to dyslipidemia.(2)
Alteration in the intestinal absorption of free fatty acidsA study observed the deficiency of essential fatty acids even in well-nourished CF patients with normal consumption of fats and in absence of clinical manifestations of poor intestinal absorption.(23) The authors believe that this problem occurs due to deficiency of micronutrients (for instance, selenium and tocopherol), chronic infection (which increases the production of eicosanoids), defect in the incorporation of fatty acids in the cell membrane or the alterations in the lipoprotein metabolism.
In another study it was observed that the alteration in the absorption of fats in CF patients occurs due to the incomplete intraluminal solubilization or reduced absorption of the long-chain fatty acids by the intestinal mucosa, without habitual insufficient enzymatic replacement.(18)
Although the deficiency of fatty acids in different cells and tissues had been initially attributed only to poor fat intestinal absorption,(24) it is now known that the disorder is primary in some patients as a result of alterations in the CFTR.(25)
Use of corticosteroidsCorticosteroids are one of the most relevant causes of secondary dyslipidemia, principally of hypertriglyceridemia.(26) As patients with CF, principally those with pulmonary complications, use these medications frequently or chronically, this can be, theoretically, one more factor contributing to dyslipidemia. However, no studies showing this association in patients with CF were found in the literature.(5)
Lipoprotein alterations in cystic fibrosisQuantitative alterationsThe principal quantitative alterations of the lipid metabolism in CF are hypertriglyceridemia and hypocholesterolemia. In patients with CF in the United States, in the 5 to 19-year age bracket, the mean serum cholesterol was 133 ± 30 mg/dL and that of TG was 126 ± 70 mg/dL in comparison with, respectively, 149 mg/dL and 55 mg/dL for the population of the same age and gender without CF. In the 20 to 44-year age bracket, the mean values for total cholesterol (TC) ad TG were 155 ± 39 mg/dL and 162 ± 118 mg/dL, when compared, respectively, with 199 mg/dL and 87 mg/dL for the control group.(5) This fact reinforces the information that CF patients present, on average, lower cholesterol values, and higher TG values in relation to their population mean. In the same study, hypertriglyceridemia defined by values above 200 mg/dL was found in only 16% of the patients.
Qualitative alterationsApproximately 85% of the patients with CF present essential fatty acid deficiency.(27) The typical profile of this deficiency is characterized by the decrease of the concentration of linoleic (18:2n-6) and docosahexaenoic acids (22:6n-3); and increase of the eicosatrienoic acid (20:3n-9), oleic acid (18:1n-9) and palmitoleic acid (16:1n-7).(27-30) As these alterations were also detected in well-nourished patients with CF and without exogenous pancreatic insufficiency and in their parents, this fact suggests that there can be a defect in the metabolism of fatty acids related to the etiology of CF, as, for instance, increase in their oxidation as a source of energy, use as precursors of the inflammatory response, defective metabolism and alterations of the CFTR.(25,27) How this profile of fatty acids is related to hypocholesterolemia and hypertriglyceridemia in patients with CF is unknown yet.
Essential fatty acids deficiency and inflammation in cystic fibrosisMorbidity and mortality in patients with CF is largely a result of pulmonary disease, which, in turn, is characterized by the increase of the inflammatory response mediated by pro-inflammatory neutrophils, lymphocytes and cytokines.(31) Patients with CF present increase of the concentration of arachidonic acid and decrease of the concentration of the docosahexaenoic acid.(24,25) The arachidonic acid is a substrate for the synthesis of prostaglandin E2, thromboxane A2 and leukotriene B4, thus contributing to the inflammatory process typical of the disease.(24) The docosahexaenoic acid is converted in potent anti-inflammatory mediators (??), so that the reduction of its serum levels is able to contribute to increase of the inflammatory response.(32) In addition, the alterations of the essential fatty acids in the CF can cause decrease of the expression and activity of the peroxisome proliferator-activated receptor alpha (PPAR-α) in lymphocytes. As the PPAR-α presents anti-inflammatory activity, its decrease, in patients with CF, could be another mechanism responsible for the exacerbated inflammatory response.(31) This observation is important since it can lead to researches which use fibrates and glitazone, synthetic activators of the PPAR-α, in the treatment of CF.
Therefore, in case it is possible to correlate the serum levels of the essential fatty acids with their tissue concentration, perhaps the attempt to normalize those with the objective of attenuating the chronic inflammatory response is reasonable. Corroborating these data, a study,(33) showed that the levels of fatty acids increased in patients with CF after treatment with antibiotics, and in another study(34) improvement of the inflammatory response in mice chronically infected by Pseudomonas aeruginosa was reported after supplementation with omega-6 fatty acids.
Dyslipidemia and cardiovascular risk in the cystic fibrosisThe role of the hypertriglyceridemia as an independent risk factor for the cardiovascular disease is still unclear.(26) In individuals without CF, this risk would be increased in situations in which the hypertriglyceridemia associates with the presence of smaller and cholesterol-rich VLDL fractions (for instance, diabetes mellitus, obesity and metabolic syndrome); and would be inexistent in hypertriglyceridemias accompanied by large and TG-rich VLDL fractions (for instance, carbohydrate-rich diets and excessive alcohol intake).(8)
As patients with CF and hypertriglyceridemia presented no atherogenic risk factors (for instance, obesity, metabolic syndrome), except for the presence of diabetes mellitus in some cases, and as the carbohydrate-rich diet does not associate with a greater risk of cardiovascular disease, it becomes even more difficult to evaluate the cardiovascular repercussion of CFD. A recent epidemiologic study showed, in the population in general, the existence of a moderate but rather significant association between elevated levels of TG and the increased risk of coronary disease.(35) This fact calls attention to the need to evaluate such risk in patients with CF and hypertriglyceridemia.
Approach to cystic fibrosis-related dyslipidemiaScreening and diagnosisThere is no specific recommendation for screening and laboratory testing diagnosis of CFD. Therefore, the orientations and reference values are extrapolated from the guidelines of the National Cholesterol Education Program, 1992(36) and from the Brazilian Cardiology Society, 2005(37) for children (Table 2); and from the recommendations of the Adult Panel III of the National Cholesterol Education Program(38) (Table 3) for adults. The reference values suggested by the Brazilian Cardiology Society for individuals aged less than 19 years differ from those of the National Cholesterol Education Program, since it proposes lower values of CT, LDL and TG.

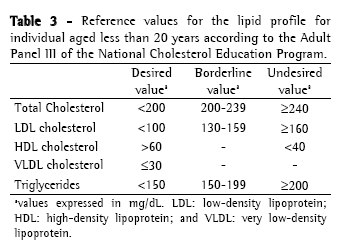
Dyslipidemia screening is carried out through the determination of the lipid profile (CT, HDL, VLDL and TG) after a 12-hour fast. The LDL is calculated using the Friedewald formula (LDL = [CT − HDL] − [TG/5]) if TGs < 400 mg/dL. Ideally, the lipid profile should be determined in individuals using their habitual diet, in absence of great recent variations in weight and without having performed vigorous physical activity or having consumed alcohol within 24 hours preceding the examination. The use of medications which alter the lipid profile such as corticosteroids, hormonal contraceptives, antihypertensives and anticonvulsants should be questioned, as well as the traditional risk factors for dyslipidemia such as smoking, hypertension and sedentary life style, among others.(26)
Abnormal results should be confirmed by a second examination. If altered test results persist, it is fundamental to exclude and treat secondary causes such as hypothyroidism, diabetes, kidney disease, sedentary life style, consumption of alcohol and use of medicaments, principally corticosteroids and estrogens.
(8,26)
Despite the frequent reports of essential fatty acids deficiency, principally the linoleic and the docosahexaenoic, there are no recommendations as to routine doses of the free fatty acids in the blood or in the membranes of the erythrocytes of the patients with CF.(30) Some of the factors which make the establishment of a normality value more difficult are: infections (decreasing the levels of free fatty acids), nutritional status, severity of the mutation of the CFTR and the fact that the serum levels of fatty acids do not reflect their tissue concentration.(25,27,33)
TreatmentNon-pharmacological treatmentDue to the greater metabolic necessity as a result of the elevated baseline energy expenditure, loss of fat due to poor intestinal absorption of fats and reduction of food intake, especially during episodes of infection, patients with CF are advised to use balanced high-protein and high-caloric diet, (120-150% of the traditional daily recommendations), with supplementation of micronutrients according to their specific deficiencies and replacement of liposoluble vitamins, as well as fibers.(2,6)
In the absence of exogenous pancreatic insufficiency, the energy recommendation should be similar to that of the population without CF, therefore the use of high-caloric diet with high level of lipids should be monitored due to the risk of dyslipidemia.(16) If there is exogenous pancreatic insufficiency, enzymatic supplementation is necessary. Normalization of fat in the stool should not be expected, since the use of excessive doses of enzymes brings no greater benefits and can cause damage to the structure of the intestine leading to a fibrosing colonopathy.(6,18) The maximum dose of pancreatic enzymes should not exceed 10,000 IU of lipase/kg/day.(39)
In patients without CF, dietetic recommendations for the treatment of hypertriglyceridemia include restriction to saturated and trans fat and increase in the consumption of omega-3 fatty acids.(26) The restriction of fats to less than 30% of the total energy consumption does not apply to CF, since the fat represents 31-35% of the caloric intake provided that, even with the pancreatic enzymes replacement, 5-20% of the fat of the diet is not absorbed.(18) In one study,(40) a greater intake of saturated and monosaturated fat and lesser intake of polyunsaturated fat has been reported in patients with CF when compared to the controls, drawing attention to the necessity of balancing the intake of fat in these patients, in order to minimize the deficiency of essential fatty acids.
Regular physical activities have been considered a fundamental part of the management of patient with CF, improving cardiorespiratory performance, muscle strength, bone mass acquisition and body perception. Exercises must be individualized, respecting the capacity and tolerance of each patient.(41)
Pharmacological treatmentThe recommendations for the treatment of hypertriglyceridemia are not as clear as for the treatment of hypercholesterolemia. Antilipidemic drugs can be recommended for severe hypertriglyceridemias (>500 or 1000 mg/dL) which do not respond to non-pharmacological treatment due to the elevated risk of pancreatitis.(8,26)
Fibrates (gemfibrozil, bezafibrate, fenofibrate) are first choice drugs for the hypertriglyceridemia treatment.(8) Its principal mechanism of action occurs through the modulation of the activity of the PPAR-α in the liver, with reduction of hepatic secretion of VLDL and TG and increase of the lipolysis of plasma TG.(26) However, its use in the pediatric age bracket awaits more experience.(37)
In relation to the other antilipidemic drugs, the statins (which reduce the hepatic production of cholesterol) and ezetimibe (which reduces the intestinal absorption of cholesterol) are not indicated, since one of the characteristics of CFD is the hypocholesterolemia. The of bile acid-sequestrant resins (colestipol and cholestyramine) are not indicated since they can increase TG levels (greater synthesis of VLDL) and they contribute to poor intestinal absorption.(37)
Some studies have been carried out with ursodeoxycholic acid with the objective of improving bile flow and thus prevent or treat liver disease related to CF and therefore, reduce the risk of dyslipidemia.(42) However, in one study,(43) no significant evidence of improvement of the hepatic function were observed with the use of such medication.
The use of supplementation with omega-3 and omega-6 fatty acids has presented conflicting results, the conduction of further studies being necessary for the recommendation of its use in the treatment of CF.(27,30,34)
Final considerations and perspectives for the futureThere are few studies evaluating the prevalence and physiopathology of CFD.
Therefore, there are no consensuses orienting on how to perform the screening for this disorder, how to prevent it and, if present, how to treat it. Due to the increase in life expectancy of the cystic-fibrotic patients and in view of the metabolic alterations presented by these individuals, it is fundamental that the conduction of prospective studies aiming to answer most of these questions, principally the role of the hypertriglyceridemia in isolation as a risk factor for cardiovascular diseases. To date, most recommendations as for the management of hypertriglyceridemia come from studies in patients without CF and with other cardiovascular risk factors. More studies aiming to better clarify the role of the deficiency of essential fatty acids in the physiopathology of the CF are also necessary.
References 1. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132(4):589-95.
2. Elborn JS. How can we prevent multisystem complications of cystic fibrosis? Semin Respir Crit Care Med. 2007;28(3):303-11.
3. Alvarez AE, Ribeiro AF, Hessel G, Bertuzzo CS, Ribeiro JD. Cystic fibrosis at a Brazilian center of excellence: clinical and laboratory characteristics of 104 patients and their association with genotype and disease severity [Article in Portuguese]. J Pediatr (Rio J). 2004;80(5):371-9.
4. Alves CA, Aguiar RA, Alves AC, Santana MA. Diabetes melito: uma importante co-morbidade da fibrose cística. J Bras Pneumol. 2007; 33(2):213-21.
5. Figueroa V, Milla C, Parks EJ, Schwarzenberg SJ, Moran A. Abnormal lipid concentrations in cystic fibrosis. Am J Clin Nutr. 2002;75(6):1005-11.
6. Dodge JA, Turck D. Cystic fibrosis: nutritional consequences and management. Best Pract Res Clin Gastroenterol. 2006;20(3):531-46.
7. Alves C, Veiga S, Souza T. Dislipidemia e risco de doença cardiovascular em crianças e adolescentes com diabetes melito tipo 1. Rev Paul Pediatria. 2007; 25(1):82-9.
8. Santos V, Canadas V, Gusmão A, Almeida NC. Investigação diagnóstica das dislipidemias. In: Vilar L, editor. Endocrinologia Clínica. Rio de Janeiro: Guanabara Koogan; 2006. p.733-47.
9. Maloy MJ, Kane JP. Disorders of lipoprotein metabolism. In: Greenspan FS, Gradner DG, editors. Basic and Clinical Endocrinology. New York: MacGraw-Hill Medical; 2004. p.766-93.
10. Bricarello SG, Bricarello LP, Alves RC. Conduta diagnóstica e terapêutica nas hiperlipidemias em Pediatria. Pediatr Mod. 1999;35(12):929-47.
11. Monte O, Cisternas JR. Dislipidemias. In: Monte O, Longui CA, Calliari LE, Kochi C, editors. Endocrinologia para o pediatra. São Paulo: Ateneu; 2006. p. 573-90.
12. Volta C, Street ME, Ziveri MA, Bonelli P, Spaggiari C, Grzincich GL, et al. Thyroid function, cytokine and IGF-IGFBP interactions in cystic fibrosis patients. Horm Res. 2005;63(4):206-10.
13. Reis FJ, Damaceno N. Fibrose cística. J Pediatr (Rio J.). 1998; 74(Supl 1):S76-S94.
14. Parks EJ, Hellerstein MK. Carbohydrate-induced hypertriacylglycerolemia: historical perspective and review of biological mechanisms. Am J Clin Nutr. 2000;71(2):412-33.
15. Frase LL, Strickland AD, Kachel GW, Krejs GJ. Enhanced glucose absorption in the jejunum of patients with cystic fibrosis. Gastroenterology. 1985;88(2):478-84.
16. Slesinski MJ, Gloninger MF, Costantino JP, Orenstein DM. Lipid levels in adults with cystic fibrosis. J Am Diet Assoc. 1994;94(4):402-8. Erratum in: J Am Diet Assoc 1994;94(10):1101.
17. Burdge GC, Goodale AJ, Hill CM, Halford PJ, Lambert EJ, Postle AD, et al. Plasma lipid concentrations in children with cystic fibrosis: the value of a high-fat diet and pancreatic supplementation. Br J Nutr. 1994;71(6):959-64.
18. Kalivianakis M, Minich DM, Bijleveld CM, van Aalderen WM, Stellaard F, Laseur M, et al. Fat malabsorption in cystic fibrosis patients receiving enzyme replacement therapy is due to impaired intestinal uptake of long-chain fatty acids. Am J Clin Nutr. 1999;69(1):127-34.
19. Fagundes ED, Silva RA, Roquete ML, Penna FJ, Reis FJ, Goulart EM, et al. Validação do escore ultra-sonográfico de Williams para o diagnóstico da hepatopatia da fibrose cística. J Pediatr (Rio J). 2004;80(5):380-6.
20. Treem WR, Stanley CA. Massive hepatomegaly, steatosis, and secondary plasma carnitine deficiency in an infant with cystic fibrosis. Pediatrics. 1989;83(6):993-7.
21. Lepage G, Yesair DW, Ronco N, Champagne J, Bureau N, Chemtob S, Bérubé D, et al. Effect of an organized lipid matrix on lipid absorption and clinical outcomes in patients with cystic fibrosis. J Pediatr. 2002;141(2):178-85.
22. Levy E, Gurbindo C, Lacaille F, Paradis K, Thibault L, Seidman E. Circulating tumor necrosis factor-alpha levels and lipid abnormalities in patients with cystic fibrosis. Pediatr Res. 1993;34(2):162-6.
23. Roulet M, Frascarolo P, Rappaz I, Pilet M. Essential fatty acid deficiency in well nourished young cystic fibrosis patients. Eur J Pediatr. 1997;156(12):952-6.
24. Strandvik B. Fatty acid metabolism in cystic fibrosis. N Engl J Med. 2004;350(6):605-7.
25. Freedman SD, Blanco PG, Zaman MM, Shea JC, Ollero M, Hopper IK, et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med. 2004;350(6):560-9.
26. Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ. 2007;176(8):1113-20.
27. Peretti N, Marcil V, Drouin E, Levy E. Mechanisms of lipid malabsorption in cystic fibrosis: the impact of fatty acids deficiency. Nut Metab [serial on the Internet]. 2005 [cited 2005 May 3];2(1):11. Available from: http://www.nutritionandmetabolism.com/content/2/1/11.
28. Christophe AB, Warwick WJ, Holman RT. Serum fatty acid profiles in cystic fibrosis patients and their parents. Lipids. 1994;29(8):569-75.
29. Farrell PM, Mischler EH, Engle MJ, Brown DJ, Lau SM. Fatty acid abnormalities in cystic fibrosis. Pediatr Res. 1985;19(1):104-9.
30. Coste TC, Armand M, Lebacq J, Lebecque P, Wallemacq P, Leal T. An overview of monitoring and supplementation of omega 3 fatty acids in cystic fibrosis. Clin Biochem. 2007;40(8):511-20.
31. Reynders V, Loitsch S, Steinhauer C, Wagner T, Steinhilber D, Bargon J. Peroxisome proliferators-activated receptor α (PPARα) down-regulation in cystic fibrosis lymphocytes. Respir Res [serial on the Internet] 2006 [cited 2006 Oct 10]; 7:104. Available from: http://respiratory-research.com/content/7/1/104.
32. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196(8):1025-37.
33. Durieu I, Abbas-Chorfa F, Drai J, Iwaz J, Steghens JP, Puget M, et al. Plasma fatty acids and lipid hydroperoxides increase after antibiotic therapy in cystic fibrosis. Eur Respir J. 2007;29(5):958-64.
34. Pierre M, Husson MO, Le Berre R, Desseyn JL, Galabert C, Béghin L, et al. Omega-3 polyunsaturated fatty acids improve host response in chronic Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1422-31.
35. Sarwar N, Danesh J, Eiriksdottir G, Sigurdsson G, Wareham N, Bingham S, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115(4):450-8.
36. National Cholesterol Education Program. Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Elk Grove Village: American Academy of Pediatrics, 1992.
37. Sociedade Brasileira de Cardiologia. I Diretriz de prevenção da aterosclerose na infância e na adolescência. Arq Bras Cardiol. 2005;85[Suppl 6]:S1-S36.
38. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-97.
39. FitzSimmons SC, Burkhart GA, Borowitz D, Grand RJ, Hammerstrom T, Durie PR, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med. 1997;336(18):1283-9.
40. Colombo C, Bennato V, Costantini D, Valmarana L, Daccò V, Zazzeron L, et al. Dietary and circulating polyunsaturated fatty acids in cystic fibrosis: are they related to clinical outcomes? J Pediatr Gastroenterol Nutr. 2006;43(5):660-5.
41. Rogers D, Prasad SA, Doull I. Exercise testing in children with cystic fibrosis. J R Soc Med. 2003;96(Suppl 43):23-9.
42. Lepage G, Paradis K, Lacaille F, Sénéchal L, Ronco N, Champagne J, et al. Ursodeoxycholic acid improves the hepatic metabolism of essential fatty acids and retinol in children with cystic fibrosis. J Pediatr. 1997;130(1):52-8.
43. Cheng K, Ashby D, Smyth R. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev. 2000;(2):CD000222.
Study carried out in the Department of Pediatric Endocrinology, Universidade Federal da Bahia - UFBA, Federal University of Bahia - School of Medicine Professor Edgard Santos University Hospital, Salvador, Brazil.
1. Coordinator of Medical Residence in Pediatric Endocrinology. Universidade Federal da Bahia - UFBA, Federal University of Bahia - School of Medicine, Salvador, Brazil.
2. Third-Year Resident in Pediatric Endocrinology. Universidade Federal da Bahia - UFBA, Federal University of Bahia - School of Medicine, Salvador, Brazil.
Correspondence to: Crésio Alves. Rua Plínio Moscoso, 222/601, CEP 40157-190, Salvador, BA, Brazil.
Tel 55 71 3357-5500. E-mail: cresio.alves@uol.com.br
Financial Support: None.
Submitted: 30 October 2007. Accepted, after review: 3 March 2008.












 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket