ABSTRACT
Objective: Mechanical ventilation (MV) itself can directly contribute to lung injury. Therefore, the aim of the present study was to investigate early biomarkers concerning oxidant/antioxidant balance, oxidative stress, and inflammation caused by short-term MV in healthy mouse lungs. Methods: Twenty male C57BL/6 mice were randomly divided into two groups: MV, submitted to low tidal volume (VT, 6 mL/kg) MV for 30 min; and spontaneous respiration (SR), used as controls. Lung homogenate samples were tested regarding the activity of various antioxidant enzymes, lipid peroxidation, and TNF-α expression. Results: In comparison with the SR group, the MV group showed a significant decrease in the activity of superoxide dismutase (≈35%; p < 0.05), together with an increase in the activity of catalase (40%; p < 0.01), glutathione peroxidase (500%; p < 0.001), and myeloperoxidase (260%; p < 0.001), as well as a reduction in the glutathione/oxidized glutathione ratio (≈50%; p < 0.05) and an increase in TNF-α expression in the MV group. Oxidative damage, assessed by lipid peroxidation, was also greater in the MV group (45%; p < 0.05). Conclusions: Our results show that short-term low VT MV can directly contribute to lung injury, generating oxidative stress and inflammation in healthy mouse lungs.
Keywords:
Lesão pulmonar induzida por ventilação mecânica; Respiração artificial; Estresse oxidativo; Inflamação; Camundongos.
RESUMO
Objetivo: A ventilação mecânica (VM) por si própria pode contribuir diretamente para a lesão pulmonar. Assim, o objetivo do presente estudo foi investigar biomarcadores precoces relacionados ao equilíbrio oxidantes/antioxidantes, estresse oxidativo e inflamação causados por VM de curta duração em pulmões de camundongos saudáveis. Métodos: Vinte camundongos C57BL/6 machos foram randomicamente divididos em dois grupos: VM, submetidos a VM com baixo volume corrente (VT, 6 mL/kg) por 30 min; e respiração espontânea (RE), utilizados como controles. Amostras de homogeneizados de pulmão foram testados quanto à atividade de enzimas antioxidantes, peroxidação lipídica e expressão de TNF-. Resultados: Comparados ao grupo RE, houve uma redução significativa na atividade de superóxido dismutase (≈35%; p < 0,05) e aumento da atividade de catalase (40%; p < 0,01), glutationa peroxidase (500%; p < 0,001) e mieloperoxidase (260%; p < 0,001), ao passo que a razão glutationa reduzida/glutationa oxidada foi menor (≈50%; p < 0,05), e houve um aumento na atividade de expressão de TNF- no grupo VM. O dano oxidativo, analisado como peroxidação lipídica, também aumentou no grupo VM (45%; p < 0.05). Conclusões: Nossos resultados demonstraram que VM de curta duração com baixa VT pode contribuir diretamente para a lesão pulmonar, gerando estresse oxidativo e inflamação em pulmões de camundongos saudáveis.
Palavras-chave:
Lesão pulmonar induzida por ventilação mecânica; Respiração artificial; Estresse oxidativo; Inflamação; Camundongos.
IntroductionOxidative stress results from an oxidant/antioxidant imbalance, favoring oxidants. The lungs have a well-developed antioxidant system in order to be protected against exposure to oxidants, which might lead to further oxidation of proteins, DNA, and lipids.(1) Key antioxidant enzymes, such as catalase, superoxide dismutase, and glutathione peroxidase, protect against superoxide- and hydrogen peroxide-induced damage. The participation of oxidative stress in lung diseases, such as COPD, acute lung injury (ALI), and asthma, has been widely demonstrated in the scientific literature.(2-4)
Recently, long-term protocols using high-stretch mechanical ventilation (MV)-i.e., with a high tidal volume (VT)-have been shown to have pro-inflammatory and oxidative effects, and they can cause lung function impairment, mainly via mechanical stress on pulmonary tissue.(5,6) For that reason, a more physiological VT (4-6 mL/kg, as suggested by the ARDS Network) has also been used; however, it has failed to reduce inflammatory response in healthy lungs.(7)
The biomarkers underlying the oxidative stress using physiological VT remain unclear. Therefore, the aim of the present study was to investigate early biomarkers concerning oxidant/antioxidant balance, oxidative stress, and inflammation caused by short-term MV in healthy mouse lungs.
MethodsAll animal procedures were performed in accordance with the Brazilian law for animal experimentation (Arouca Law no. 11.794, October 8, 2008) and were approved by the Rio de Janeiro State University Committee for the Ethical Care and Use of Laboratory Animals. We purchased twenty C57BL/6 male mice (8 weeks old; weight, 20-24 g) from the Veterinary Institute of Fluminense Federal University, located in Niterói, Brazil. The animals were randomly divided into two groups (n = 10/group): MV; and spontaneous respiration (SR).
The animals were sedated with diazepam (1 mg/kg, i.p.), anesthetized with pentobarbital (20 mg/kg, i.p.), and submitted to tracheal intubation. Those in the MV group were then subjected to MV with a constant flow ventilator (Samay VR15, Universidad de la Republica, Montevideo, Uruguay). The animals in the SR group underwent sham procedures (sedation, anesthesia, and tracheal intubation) but were not submitted to MV. In accordance with the protocol, VT was set at 6 mL/kg, with respiratory flow of 1 mL/s, FiO2 of 0.21 (with dry gas), RR of 100 breaths/min, and positive end-expiratory pressure of 3 cmH2O.(8) The mice were ventilated with room air. After 30 min of MV or SR, the mice were immediately sacrificed.
Airspaces were washed with buffered saline solution (500 µL) three consecutive times (final volume, 1.2-1.5 mL). The fluid was withdrawn and stored on ice. Total mononuclear and polymorphonuclear cell numbers were determined with a particle counter (Z1; Beckman Coulter, Carlsbad, CA, USA). Differential cell counts were performed on cytospin preparations (Shandon, Waltham, MA, USA) stained with Diff-Quik (Baxter Dade, Dudingen, Switzerland). At least 200 cells per BAL fluid (BALF) sample were counted using standard morphological criteria.
The right ventricle was perfused with saline to remove blood. Lungs were removed and then homogenized on ice with 10% (w/v) 0.1 M potassium phosphate + 5 mM EDTA buffer solution (pH, 7.3) using an Ultra-Turrax T8 homogenizer (IKA, Toronto, Ontario, Canada) and then centrifuged at 3,000 g for 5 min. Supernatants were stored at 20°C for subsequent protein measurements. The total protein content in the samples from BALF and lung homogenates was determined by the Bradford protein assay.
In order to determine whether MV resulted in redox imbalance, catalase, superoxide dismutase, glutathione peroxidase, reduced glutathione (GSH), and oxidized glutathione (GSSG) were assayed spectrophotometrically in the lung homogenates. Catalase activity was measured by the rate of decrease in H2O2 concentration at 240 nm.(9) Superoxide dismutase activity was assayed by measuring the inhibition of adrenaline auto-oxidation as absorbance at 480 nm.(10) Glutathione peroxidase activity was measured by monitoring the oxidation of NADPH at 340 nm in the presence of H2O2.(11) Lung homogenates were acidified with 5% 5-sulfosalicylic acid (1:1) for GSH and GSSG analyses.(12)
Malondialdehyde concentration was measured by using the thiobarbituric acid reactive substances (TBARS) method.(13) Samples of organ homogenates (400 µL) were deproteinized with 800 µL of 10% trichloroacetic acid and centrifuged at 900 g for 10 min. After that, 1,000 µL of supernatants were mixed with 1,000 µL of 0.67% TBARS. The mixture was heated for 60 min in boiling water and then cooled. Absorbance of the organic phase containing the pink chromogen was measured spectrophotometrically at 532 nm. The method was standardized with increasing concentrations of malondialdehyde. Malondialdehyde equivalents were expressed in nmol/mg of protein.
Myeloperoxidase activity was measured by using H2O2, hexadecyltrimethylammonium bromide (HTAB) and 3,3',5,5' tetramethylbenzidine (TMB).(14) Initially, 100 µL of organ homogenate samples were centrifuged with 900 µL of HTAB at 14,000 g for 15 min. After that, 75 µL of the supernatant was incubated with 5 µL of TMB for 5 min at 37°C. The mixture was then incubated with 50 µL of H2O2 for 10 min at 37°C, after which 125 µL of sodium acetate buffer was added. The reaction was read in a microplate reader (model 550; Bio-Rad, Hercules, CA, USA) at 630 nm. The method was standardized with increasing concentrations of myeloperoxidase. Myeloperoxidase activity was expressed in mU/mg of protein.
Proteins from tissue homogenates were separated by PAGE and transferred to nitrocellulose membranes (100 V for 3 h at 4°C). Membranes were then washed and blocked in PBS-Tween buffer containing 5% skimmed milk and 0.05% Tween for 2 h and subsequently incubated for TNF- detection using specific primary goat anti-mouse antibody (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were then incubated with horseradish peroxidase-antibody conjugate (1:2000) directed against the primary antibody for 2 h. Membranes were then treated with chemiluminescent agents (luminol and enhancer, ECL Plus; Amersham Biosciences, Piscataway, NJ, USA) and exposed to light-sensitive film. Ponceau stain was used as a loading control.
The normality of all data was tested using the Kolmogorov-Smirnov test. Data were expressed as means ± SE of the mean. Parametric data were compared using unpaired Student's t-test. Non-parametric data were compared using the Wilcoxon test. Values of p < 0.05 were considered significant.
Results
We first tested whether MV for 30 min triggered an inflammatory response in mouse lungs. There was a significant increase in TNF- expression in the lung homogenates after 30 min of MV (Figure 1). In addition, we measured myeloperoxidase activity, which is the resultant of the activation of neutrophils (Table 1). There was a four-fold increase in myeloperoxidase activity in the MV group in comparison with the SR group (p < 0.001).
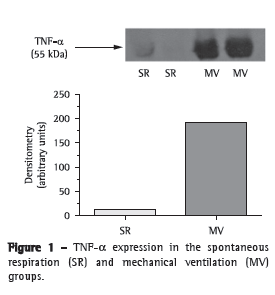
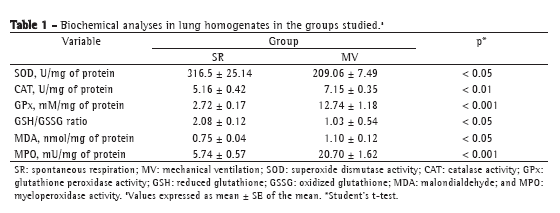
After 30 min of MV, there was a two-fold increase in total protein concentration (0.117 ± 0.010 g/dL vs. 0.06 ± 0.01 g/dL in the SR group; p < 0.01) in BALF; however, no alterations were found in tissue homogenates (SR group: 0.23 ± 0.05 g/dL, and MV group: 0.22 ± 0.05 g/dL; p > 0.05). After that, we measured antioxidant activity in order to determine whether MV produced redox imbalance in lung tissue (Table 1). In comparison with the SR group, the MV group showed significantly lower superoxide dismutase activity (p < 0.05), significantly increased catalase activity (p < 0.01), a five-fold increase in glutathione peroxidase activity (p < 0.001), a 50% decrease in the GSH/GSSG ratio (p < 0.05), and a 45% increase in malondialdehyde levels (p < 0.05).
DiscussionWe demonstrated that 30 min of MV using a VT of 6.0 mL/kg caused the release of TNF-, lipid peroxidation, and oxidative stress in healthy mouse lungs. To our knowledge, this is the first study describing the early inflammatory markers and redox imbalance markers with the use of a more physiological VT in the lungs of healthy mice.
The damaging effects of MV due to mechanical forces have been described in the literature. Studies have shown that sheared endothelial cells produce inflammatory mediators, including the release of nuclear factor kappa B-mediated TNF-,(15) and that mechanotransduction mechanisms, in which the cytoskeleton transmits tensions directly to internal organelles, are involved in producing reactive oxygen species (ROS) and signaling adhesion proteins, such as P-selectin.(16,17) Although the mechanisms that underlie the mechanotransduction that induces ROS production in the lungs still need to be elucidated, increased mitochondrial NADPH oxidase activity has been related to ROS production in endothelial cells as a response to mechanical stretch, signaling the activation of inflammatory responses.(18)
It is widely known that TNF- is an early inflammatory cytokine that subsequently regulates early neutrophil infiltration and eosinophil recruitment into the lung and airspace, produced by other inflammatory cells, such as macrophages, or via mechanotransduction mechanisms.(19) In previous studies, it has been reported that TNF- levels were unchanged following 2 h of MV.(20) In view of the fact that TNF- expression was significantly increased in the lung homogenates of the MV group, we believe that short-term MV with a low VT was able to initiate the inflammatory response in the mouse lungs. The elevated protein content in BALF and the increase in myeloperoxidase activity(21) might support this statement regarding our MV group. We believe that the use of a distinct mouse strain might account for these differences, since we have shown, in a previous study, that C57BL/6 mice are more susceptible to oxidative damage than are Swiss mice.(4)
It has been previously reported that MV is a modulator of oxidative stress not only in the lungs,(22) but also in other organs,(23) and that nuclear factor (erythroid-derived-2)-related factor-2 (NRF-2), which is a signal-activated transcription factor for antioxidant enzymes, plays a critical role in that stress.(24) We believe that, in the present study, two main processes were involved in the lung injury caused by MV: oxidative stress and inflammation. It is known that MV increases neutrophil recruitment and myeloperoxidase activity in animals and humans.(25) Myeloperoxidase is mainly released by activated neutrophils, and this is also characterized by powerful pro-oxidative and pro-inflammatory properties. In addition, myeloperoxidase catalyzes the production of potent oxidants, such as hypochlorous acid, and of the nitrating species (such as nitrogen dioxide from nitrite).(26) Therefore, oxidative damage is most likely to occur in an environment where myeloperoxidase activity is high.
The association between lipid oxidation in the lungs and MV has been previously reported.(27) In the present study, the amount of peroxidation-derived aldehydes was increased in the lung homogenates after 30 min of MV. Due to their stability, lipid peroxidation-derived aldehydes can diffuse within or even escape from the cell and attack targets far from the site of the original free radical event.
Because there were no gross histological alterations in the alveolar-capillary membrane in low VT models of MV (data not shown), we suggest that mechanical stress-induced alveolar cell recruitment and decreased tissue oxygenation levels might have played important roles in the early oxidative stress process in our model. Regarding pro-oxidant mechanisms, it has been shown that under hypoxic or shear stressed conditions, lung cells respond to damaging stimuli by producing ROS, such as superoxide (O2−), either via NADPH or via increases in xanthine oxidase levels, leading to the formation of peroxynitrite (ONOO−) if nitric oxide is present in the environment, mainly in components of the alveolar-capillary membrane.(28)
We found a significant decrease in the superoxide dismutase activity in the MV group. The reduction of superoxide dismutase activity might be in part due to the loss of enzyme specific activity, as observed by the action of oxidants. Therefore, we suggest that reduced superoxide dismutase activity might have a direct effect on O2− overproduction in the lungs. However, O2− might have first been converted into ONOO−, since the rate constant is over 3.5 times faster than is that of the dismutation of O2− by superoxide dismutase.(29) In addition, in the present study, increased H2O2 production might have served mainly as a substrate for myeloperoxidase, rather than being totally inactivated by catalase, as indicated by the reduction of catalase activity in the MV group. Furthermore, our data indicate a possible lipid peroxidation-inducing effect of ROS, which might have resulted in increased malondialdehyde levels in the MV group. In order to buffer the oxidative action of reactive lipids, glutathione peroxidase activity also increases and, in turn, the GSH/GSSG ratio decreases.
Short-term, low-VT MV induced not only inflammation, but also redox imbalance and oxidative damage in healthy mouse lungs. Although our model offers opportunities to study the early biomarkers of oxidative response that lay behind MV, it also presents limitations. One cannot exclude the possibility that the oxidative stress status might be transient in this model and might therefore not necessarily result in histoarchitectural changes in the mouse lungs. In addition, the repair mechanism and antioxidants might be simultaneously activated to counteract the intense increases in myeloperoxidase and TNF- in order to achieve a new redox balance before an alteration of the 'normal' function/morphology could be detected. The pro-inflammatory status was analyzed only by quantifying TNF- expression. The other cytokines and regulators of inflammation, such as nuclear factor kappa B and activator protein 1, should be considered in future studies. Furthermore, NRF-2, which is responsible for the release of antioxidant enzymes and the regulation of redox imbalance, should be studied in more detail. In the present study, humidified air was not used, and neither PaO2 nor blood pressure was measured. Therefore, some of the inflammatory and oxidative stress parameters analyzed could be the result of hypoxia related to MV efficiency. However, various authors speculated that atelectrauma might play a role in ventilator-induced lung injury (VILI) due to low VT, since parts of the lung might undergo various hypoxemic periods during MV.(30) Therefore, it is possible that low VT resulted in inadequate ventilation volumes, causing tissue hypoxia. With a low VT, it is possible that gas exchange is hampered because of dead space ventilation. Small volumes might not lead to adequate exchange of air in the alveoli if they are close to the dead space. Although we cannot rule out the possibility that the low VT led to hypoxia, mostly due to dead space ventilation, we do not think that this was the main reason for oxidative stress and inflammation. Other problems related to hypoxia include low FiO2 and the use of non-humidified air. Taken together, these limitations do not invalidate our results but rather open the field of study regarding oxidative stress and inflammation during MV in mouse lungs.
We cannot state that there is a direct cause-and-effect relationship between our model of VILI and oxidative stress/inflammation. It is possible that MV induces a process of mechanical stress, which leads to oxidative stress and inflammation. This should be investigated in future studies. However, health professionals should be aware of the fact that early lung-protective ventilation used as a therapeutic approach might also cause an early oxidative stress response. In addition, the modulation of the early inflammatory and oxidative response might provide strategies for reducing VILI.
References1. Andrade Jr DR, Souza RB, Santos SA, Andrade DR. Oxygen free radicals and pulmonary disease. J Bras Pneumol. 2005;31(1):60-8.
2. Park CS, Kim TB, Lee KY, Moon KA, Bae YJ, Jang MK, et al. Increased oxidative stress in the airway and development of allergic inflammation in a mouse model of asthma. Ann Allergy Asthma Immunol. 2009;103(3):238-47.
3. Syrkina O, Jafari B, Hales CA, Quinn DA. Oxidant stress mediates inflammation and apoptosis in ventilator-induced lung injury. Respirology. 2008;13(3):333-40.
4. Rueff-Barroso CR, Trajano ET, Alves JN, Paiva RO, Lanzetti M, Pires KM, et al. Organ-related cigarette smoke-induced oxidative stress is strain-dependent. Med Sci Monit. 2010;16(7):BR218-26.
5. Wilson MR, Choudhury S, Goddard ME, O'Dea KP, Nicholson AG, Takata M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J Appl Physiol. 2003;95(4):1385-93.
6. Taniguchi LU, Caldini EG, Velasco IT, Negri EM. Cytoskeleton and mechanotransduction in the pathophysiology of ventilator-induced lung injury. J Bras Pneumol. 2010;36(3):363-71.
7. Wolthuis EK, Vlaar AP, Choi G, Roelofs JJ, Juffermans NP, Schultz MJ. Mechanical ventilation using non-injurious ventilation settings causes lung injury in the absence of pre-existing lung injury in healthy mice. Crit Care. 2009;13(1):R1.
8. Pires KM, Bezerra FS, Machado MN, Zin WA, Porto LC, Valença SS. N-(2-mercaptopropionyl)-glycine but not allopurinol prevented cigarette smoke-induced alveolar enlargement in mouse. Respir Physiol Neurobiol. 2011;175(3):322-30.
9. Aebi H. Catalase in vitro. Methods Enzymol. 1984;105:121-6.
10. Bannister JV, Calabrese L. Assays for superoxide dismutase. Methods Biochem Anal. 1987;32:279-312.
11. Flohé L, Günzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114-21.
12. Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1(6):3159-65.
13. Draper HH, Hadley M. Malondialdehyde determination as index of lipid peroxidation. Methods Enzymol. 1990;186:421-31.
14. Krueger AJ, Yang JJ, Roy TA, Robbins DJ, Mackerer CR. An automated myeloperoxidase assay. Clin Chem. 1990;36(1):158.
15. Rich PB, Douillet CD, Hurd H, Boucher RC. Effect of ventilatory rate on airway cytokine levels and lung injury. J Surg Res. 2003;113(1):139-45.
16. Ichimura H, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest. 2003;111(5):691-9.
17. Hsieh HJ, Cheng CC, Wu ST, Chiu JJ, Wung BS, Wang DL. Increase of reactive oxygen species (ROS) in endothelial cells by shear flow and involvement of ROS in shear-induced c-fos expression. J Cell Physiol. 1998;175(2):156-62.
18. De Keulenaer GW, Chappell DC, Ishizaka N, Nerem RM, Alexander RW, Griendling KK. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state: role of a superoxide-producing NADH oxidase. Circ Res. 1998;82(10):1094-101.
19. Bhattacharya S, Sen N, Yiming MT, Patel R, Parthasarathi K, Quadri S, et al. High tidal volume ventilation induces proinflammatory signaling in rat lung endothelium. Am J Respir Cell Mol Biol. 2003;28(2):218-24.
20. Pedreira PR, García-Prieto E, Parra D, Astudillo A, Diaz E, Taboada F, et al. Effects of melatonin in an experimental model of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295(5):L820-7.
21. Brégeon F, Roch A, Delpierre S, Ghigo E, Autillo-Touati A, Kajikawa O, et al. Conventional mechanical ventilation of healthy lungs induced pro-inflammatory cytokine gene transcription. Respir Physiol Neurobiol. 2002;132(2):191-203.
22. Whidden MA, McClung JM, Falk DJ, Hudson MB, Smuder AJ, Nelson WB, et al. Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol. 2009;106(2):385-94.
23. Douillet CD, Robinson WP 3rd, Zarzaur BL, Lazarowski ER, Boucher RC, Rich PB. Mechanical ventilation alters airway nucleotides and purinoceptors in lung and extrapulmonary organs. Am J Respir Cell Mol Biol. 2005;32(1):52-8.
24. Papaiahgari S, Yerrapureddy A, Reddy SR, Reddy NM, Dodd-O JM, Crow MT, et al. Genetic and pharmacologic evidence links oxidative stress to ventilator-induced lung injury in mice. Am J Respir Crit Care Med. 2007;176(12):1222-35.
25. Erdbrügger U, Hellmark T, Bunch DO, Alcorta DA, Jennette JC, Falk RJ, et al. Mapping of myeloperoxidase epitopes recognized by MPO-ANCA using human-mouse MPO chimers. Kidney Int. 2006;69(10):1799-805.
26. Kumar AP, Ryan C, Cordy V, Reynolds WF. Inducible nitric oxide synthase expression is inhibited by myeloperoxidase. Nitric Oxide. 2005;13(1):42-53.
27. Ben Baouali A, Aube H, Maupoil V, Blettery B, Rochette L. Plasma lipid peroxidation in critically ill patients: importance of mechanical ventilation. Free Radic Biol Med. 1994;16(2):223-7.
28. Singh JM, Stewart TE. High-frequency mechanical ventilation principles and practices in the era of lung-protective ventilation strategies. Respir Care Clin N Am. 2002;8(2):247-60.
29. Hauber HP, Karp D, Goldmann T, Vollmer E, Zabel P. Effect of low tidal volume ventilation on lung function and inflammation in mice. BMC Pulm Med. 2010;10:21.
30. Nerlich S. Critical care management of the patient with acute respiratory distress syndrome (ARDS). Part 1: Pathophysiology and implications for mechanical ventilation. Aust Crit Care. 1997;10(2):49-54.
Study carried out at the Evandro Chagas Clinical Research Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil.
Correspondence to: Renata de Lima Orofino. Rua J G de Araujo Jorge, 36, Casa 2, Condomínio Giardini di Milano, Recreio dos Bandeirantes, CEP 22790-689, Rio de Janeiro, RJ, Brasil.
Tel. 55 21 3865-9601. E-mail: renataorofino@yahoo.com.br
Financial support: None.
Submitted: 30 June 2011. Accepted, after review: 5 December 2011.
About the authorsRenata de Lima Orofino
Physician. Laboratory for Clinical Research on Mycobacterial Disease, Evandro Chagas Clinical Research Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil.
Pedro Emmanuel Americano do Brasil
Infectious Disease Physician. Evandro Chagas Clinical Research Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil.
Anete Trajman
Coordinator of the Professional Master's Program in Health Education. Gama Filho University, Rio de Janeiro, Brazil; and Adjunct Professor. McGill University, Montreal, Canada.
Carolina Arana Stanis Schmaltz
Physician. Laboratory for Clinical Research on Mycobacterial Disease, Evandro Chagas Clinical Research Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil.
Margareth Dalcolmo
Physician. Outpatient Clinic of the Hélio Fraga Referral Center for Tuberculosis, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil.
Valéria Cavalcanti Rolla
Head. Laboratory for Clinical Research on Mycobacterial Disease, Evandro Chagas Clinical Research Institute, Oswaldo Cruz Foundation, Rio de Janeiro, Brazil.












 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket