ABSTRACT
Objective: To investigate the effect of intermittent hypoxia-a model of obstructive sleep apnea (OSA)-on pancreatic expression of uncoupling protein-2 (UCP2), as well as on glycemic and lipid profiles, in C57BL mice. Methods: For 8 h/day over a 35-day period, male C57BL mice were exposed to intermittent hypoxia (hypoxia group) or to a sham procedure (normoxia group). The intermittent hypoxia condition involved exposing mice to an atmosphere of 92% N and 8% CO2 for 30 s, progressively reducing the fraction of inspired oxygen to 8 ± 1%, after which they were exposed to room air for 30 s and the cycle was repeated (480 cycles over the 8-h experimental period). Pancreases were dissected to isolate the islets. Real-time PCR was performed with TaqMan assays. Results: Expression of UCP2 mRNA in pancreatic islets was 20% higher in the normoxia group than in the hypoxia group (p = 0.11). Fasting serum insulin was higher in the hypoxia group than in the normoxia group (p = 0.01). The homeostasis model assessment of insulin resistance indicated that, in comparison with the control mice, the mice exposed to intermittent hypoxia showed 15% lower insulin resistance (p = 0.09) and 21% higher pancreatic β-cell function (p = 0.01). Immunohistochemical staining of the islets showed no significant differences between the two groups in terms of the area or intensity of α- and β-cell staining for insulin and glucagon. Conclusions: To our knowledge, this is the first report of the effect of intermittent hypoxia on UCP2 expression. Our findings suggest that UCP2 regulates insulin production in OSA. Further study of the role that UCP2 plays in the glycemic control of OSA patients is warranted.
Keywords:
Blood glucose; Sleep apnea syndromes; Pancreas; Glucagon-secreting cells.
RESUMO
Objetivo: Investigar o efeito da hipóxia intermitente com um modelo de apneia obstrutiva do sono (AOS) sobre a expressão de uncoupling protein-2 (UCP2), assim como sobre perfis glicêmicos e lipídicos, em camundongos C57BL. Métodos: Camundongos C57BL machos foram expostos a hipóxia intermitente ou hipóxia simulada (grupo controle) 8 h/dia durante 35 dias. A condição de hipóxia intermitente envolveu a exposição dos camundongos a uma atmosfera de 92% de N e 8% de CO2 por 30 s, com redução progressiva de fração de O2 inspirado até 8 ± 1%, seguida por exposição a ar ambiente por 30 s e repetições do ciclo (480 ciclos no período experimental de 8 h). Os pâncreas foram dissecados para isolar as ilhotas. Foi realizada PCR em tempo real utilizando o método TaqMan. Resultados: A expressão do mRNA da UCP2 nas ilhotas pancreáticas foi 20% maior no grupo controle que no grupo hipóxia (p = 0,11). A insulina sérica de jejum foi maior no grupo hipóxia do que no grupo controle (p = 0,01). O modelo de avaliação da homeostase de resistência à insulina indicou que, em comparação com os camundongos controle, aqueles expostos à hipóxia intermitente apresentaram 15% menor resistência à insulina (p = 0,09) e 21% maior função das células beta (p = 0,01). A coloração das ilhotas pancreáticas por imuno-histoquímica não mostrou diferenças significativas entre os grupos em termos da área ou da intensidade das células alfa e beta, marcadas por insulina e glucagon. Conclusões: Segundo nosso conhecimento, esta é a primeira descrição do efeito da hipóxia intermitente sobre a expressão da UCP2. Nossos achados sugerem que UCP2 regula a produção de insulina na AOS. Futuras investigações sobre o papel da UCP2 no controle glicêmico em pacientes com AOS são justificadas.
Palavras-chave:
Glicemia; Síndromes da apneia do sono; Pâncreas; Células secretoras de glucagon. Abstract
IntroduçãoA apneia obstrutiva do sono (AOS) é caracterizada pelo colapso recorrente da faringe durante o sono. Cada evento apneico resulta em dessaturação de oxigênio e, ocasionalmente, em microdespertar, causando hipóxia intermitente, estresse oxidativo e fragmentação do sono.(1) Até 32% da população adulta é acometida pela AOS.(2)
Diversos desfechos cardiovasculares e metabólicos têm sido atribuídos à AOS.(3) A síndrome metabólica compartilha várias características com a AOS, incluindo obesidade, hiperlipidemia, hipertensão e resistência à insulina, que podem estar implicadas em maior risco de doença cardiovascular.(4-6) Em pacientes com AOS, pode-se melhorar a síndrome metabólica por meio de tratamento com pressão positiva contínua nas vias aéreas,(7-9) indicando uma relação causal entre as duas condições.
Modelos experimentais que submetem animais à hipóxia intermitente são utilizados para estudar as consequências da AOS. Há relatos de resistência à insulina e de alterações metabólicas após exposição a esses modelos.(10,11) Os mecanismos subjacentes às alterações induzidas pela hipóxia intermitente no metabolismo da glicose ainda não foram totalmente explorados.
A uncoupling protein-2 (UCP2) é um regulador negativo da secreção de insulina das células beta pancreáticas.(12-14) As UCPs, localizadas na membrana mitocondrial interna, translocam prótons do espaço intermembranar para a matriz mitocondrial. Os prótons que vazam através de uma UCP não podem mais ser utilizados para conduzir a rotação da ATP sintase, e, assim, esse vazamento de prótons diminui a geração de ATP. Ao reduzir a síntese de ATP, a UCP2 regula a secreção de insulina estimulada por glicose.(15) Os radicais livres gerados durante a hipóxia-reoxigenação na apneia do sono podem desempenhar um papel regulador nas células beta pancreáticas.(16) Essa é a fundamentação para a investigação de uma possível relação apneia-UCP2.
Em virtude das proporções epidêmicas que o diabetes e a AOS estão assumindo, há interesse em entender a interação entre essas duas condições. Em uma extensa pesquisa da literatura, não encontramos nenhuma referência à regulação molecular da secreção de insulina na AOS. Nossa hipótese era que a hipóxia intermitente alteraria a expressão da UCP2. Portanto, investigamos o efeito da hipóxia intermitente na expressão da UCP2 pancreática em camundongos C57BL.
MétodosAnimais e hipóxia intermitenteForam avaliados 36 camundongos C57BL machos, em dois grupos: um grupo exposto a hipóxia intermitente (grupo hipóxia, n = 18) e outro submetido a hipóxia simulada (grupo controle, n = 18). Todos os camundongos tinham 8-9 semanas de idade no início do estudo. Todos os procedimentos foram realizados de acordo com o Guia para o Cuidado e Uso de Animais de Laboratório,(17) e o protocolo do estudo foi aprovado pelo Comitê de Ética em Pesquisa do Hospital de Clínicas de Porto Alegre, em Porto Alegre, RS (Protocolo nº 09-300).
Grupos de 6 camundongos permaneceram alojados em condições de temperatura controlada (22,5-24,5°C), em ciclo claro-escuro de 12 h (luzes acesas às 7h00), e receberam ração padrão para camundongos e água à vontade. Utilizando uma balança de precisão com resolução de 0,01 g (AS 5500C; Marte Científica, São Paulo, Brasil), pesamos os camundongos no início do estudo e, posteriormente, a cada 3 dias, até o final do experimento. As ração padrão em cada gaiola foram pesadas diariamente, para quantificação da ingestão de alimentos.
Ao longo de 35 dias, durante 8 h por dia no período de luzes acesas (das 9h00 às 17h00), os camundongos foram colocados no sistema de hipóxia intermitente. Na condição de hipóxia intermitente, uma mistura de gás composta por 92% de nitrogênio e 8% de CO2 foi administrada na câmara de hipóxia por 30 s, progressivamente reduzindo a fração inspirada de oxigênio (FiO2) de 21% para um nadir de 8 ± 1%. Em seguida, um ventilador moveu ar ambiente para dentro da câmara por 30 s, restaurando a FiO2 para 21% em 5 s. Portanto, cada ciclo de hipóxia/normoxia teve a duração de 60 s (60 ciclos/h), para um total de 480 ciclos de hipóxia durante o período de 8 h. Na condição controle (normoxia; hipóxia simulada), os camundongos permaneceram alojados em uma gaiola adjacente e foram expostos aos mesmos ciclos de ventilador desligado/ventilador ligado, embora nenhuma mistura gasosa tenha sido administrada na câmara em momento algum.
No final do período experimental, os animais foram submetidos a jejum de 6-10 h e, em seguida, foram anestesiados por injeção intraperitoneal de cetamina (100 mg/kg) e xilazina (10 mg/kg). Após a confirmação da anestesia, amostras de sangue foram coletadas do seio retro-orbital. A sangria dos camundongos foi realizada durante o procedimento. As amostras de sangue foram centrifugadas a 10.000 rpm por 10 min a 4°C para obtenção das amostras de soro. Os pâncreas de 12 camundongos por grupo foram dissecados, extraídos, embebidos por 24 h em cinco volumes de reagente de armazenamento de tecido (RNAlater®; Applied Biosystems/Ambion, Austin, TX, EUA) e armazenados a 4°C por toda a noite, sendo em seguida congelados a −80°C até a análise. Os pâncreas de 6 camundongos por grupo foram fixados em formalina a 10% por 24 h e incluídos em parafina para processamento futuro.
PCR em tempo real para UCP2, imuno-histoquímica das ilhotas e ensaios bioquímicos
Os pâncreas foram descongelados, dissecados para isolar as ilhotas e homogeneizados em solução otimizada à base de guanidina/fenol para lise. O RNA total foi isolado utilizando-se solução reagente TRI (Applied Biosystems/Ambion). A quantificação e teste de qualidade do RNA foram realizados por medição fotométrica em espectrofotômetro (ND-1000; NanoDrop Technologies, Wilmington, DE, EUA). Utilizando-se um kit de transcrição reversa High Capacity cDNA (Applied Biosystems, Foster City, CA, EUA), cDNA foi sintetizado a partir de 2 μg de RNA altamente purificado (A260/A280 > 1,95). O cDNA foi armazenado a −20°C e diluído em água livre de DNase (1:100) antes da quantificação relativa por PCR em tempo real. A análise de expressão gênica foi realizada utilizando-se o método TaqMan para o gene UCP2 (Mm00627598_m1; Applied Biosystems), com as seguintes sequências de primers: forward, 5′-ACAAGACCATTGCACGAGAG-3′ e reverse, 5′ ATGAGGTTGGCTTTCAGGAG-3′. Os resultados foram normalizados para expressão de gliceraldeído-3-fosfato desidrogenase (GAPDH, do inglês glyceraldehyde-3-phosphate dehydrogenase). A ciclagem térmica foi realizada em sistema de PCR em tempo real (StepOneT; Applied Biosystems).
A expressão gênica foi quantificada utilizando-se o método 2-ΔΔCt (ciclo limiar). (18) Cada amostra foi analisada em duplicata, e o valor de ΔCt foi obtido subtraindo-se o valor de Ct do GAPDH do valor de Ct do gene UCP2. Para o cálculo da diferença entre os grupos, a média do valor de ΔCt obtida para o grupo controle foi utilizada para o cálculo de ΔΔCtdo gene (2-ΔΔCt).
Os pâncreas incluídos em parafina foram cortados em secções de 5 µm. As secções foram montadas em lâminas de vidro, revestidas com 3-aminopropiltrioetoxisilano, desparafinizadas em xileno (duas mudanças, 5 min cada) e reidratadas em uma série graduada de etanol (quatro lavagens sucessivas), sendo em seguida imersas em água destilada e PBS. Para a recuperação antigênica, as lâminas foram imersas em tampão citrato de sódio a 10 mM, pH 6,0, aquecidas em micro-ondas por 21 min e resfriadas até a temperatura ambiente. Para inibição da atividade da peroxidase endógena, as lâminas foram imersas duas vezes em peróxido de hidrogênio a 5% por 15 min. Os sítios não específicos de ligação de anticorpos foram bloqueados com leite em pó desnatado a 5% diluído em PBS por 20 min. A marcação imuno-histoquímica para células beta e alfa foi realizada utilizando-se o anticorpo policlonal anti-insulina de porquinho-da-índia (A0564, diluição de 1:200; Dako, Carpinteria, CA, EUA) e o anticorpo policlonal anti-glucagon de coelho (GA 1181, 1:4.000; Enzo Life Sciences, Farmingdale, NY, EUA), respectivamente. Após três lavagens, em água corrente, água destilada e PBS, respectivamente, as lâminas foram incubadas por 1 h em câmara de umidade à temperatura ambiente. Foram então lavadas uma vez com água destilada e três vezes com PBS e, em seguida, foram incubadas com um anticorpo secundário (Picture-MAX Polymer Kit; Invitrogen, Camarillo, CA, EUA) por 30 min na câmara de umidade. A revelação das lâminas foi realizada utilizando-se 3,3'-diaminobenzidina como cromógeno, de acordo com as instruções do fabricante. As lâminas foram então contracoradas com hematoxilina de Harris por 20 s, lavadas em água corrente e enxaguadas com hidróxido de amônio para se obter uma cor azul clara. Em seguida, foram lavadas uma vez com água, duas vezes com etanol e três vezes com xilol. Todas as secções foram analisadas em microscópio de luz (aumento, 400×), digitalizadas e avaliadas utilizando-se um sistema de análise de imagem por computador (Image-Pro Plus, version 6.0; Media Cybernetics, Bethesda, MD, EUA). A área de células beta nas ilhotas foi determinada pela área celular positiva para insulina, que foi calculada como a razão entre a área intrailhota positiva para insulina e a área total da ilhota. A área relativa do glucagon, que representava a quantidade de células alfa em cada ilhota, foi avaliada pelo mesmo método. Os índices de células positivas para insulina e de células positivas para glucagon para cada grupo foram calculados como a média de pelo menos 24 secções. Os núcleos não foram levados em consideração para a análise da composição celular das ilhotas.
Os níveis de glicemia de jejum, colesterol total, HDL colesterol e triglicérides foram medidos por métodos enzimáticos colorimétricos utilizando-se kits disponíveis comercialmente (Labtest Diagnostics, Belo Horizonte, Brasil). Os níveis de LDL colesterol foram estimados pela fórmula de Friedewald(19):
LDL colesterol = (colesterol total - HDL colesterol − triglicérides) / 5
A insulina sérica de jejum foi determinada utilizando-se um kit ELISA insulina de camundongo (ALPCO Diagnostics, Salem, NH, EUA). O glucagon sérico de jejum foi quantificado utilizando-se um kit de imunoensaio enzimático (EIA; Phoenix Pharmaceuticals, Burlingame, CA). Para a avaliação do grau de resistência à insulina, foi calculado o valor do modelo de avaliação da homeostase da resistência à insulina (HOMA-IR, do inglês homeostasis model assessment of insulin resistance) utilizando-se a seguinte fórmula(20):
HOMA-IR = insulina sérica de jejum (µU/mL) × glicemia de jejum (mmol/L) / 22,5
Além disso, calculou-se o valor do HOMA da função das células beta pancreáticas (HOMA-β) da seguinte maneira:
HOMA-β = [nível de insulina sérica de jejum (µU/mL) × 20] / [nível de glicemia de jejum (mmol/L) - 3,5]
Análise estatísticaOs dados foram tabulados e expressos em medianas e intervalos interquartis e em gráficos box plot mostrando os percentis 10, 25, 50, 75 e 90, assim como os valores aberrantes. O teste U de Mann-Whitney foi utilizado para comparação entre grupos em relação à mediana do mRNA da UCP2, à área celular positiva para insulina/glucagon e aos valores dos doseamentos bioquímicos. O peso corporal em cada grupo foi ajustado para ingestão de alimentos utilizando-se o método das equações de estimação generalizadas (EEG), seguido por comparações pareadas com correção sequencial de Bonferroni. Todas as análises estatísticas foram realizadas com o Statistical Package for the Social Sciences, versão 17.0 para Windows (SPSS Inc., Chicago, IL, EUA). A significância estatística foi assumida se a probabilidade de erro alfa fosse < 0,05.
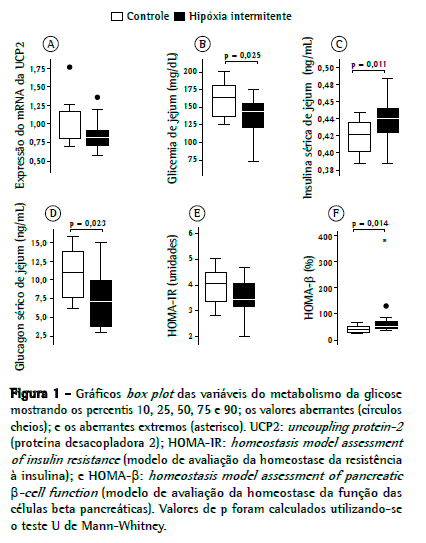
ResultadosOs dados mostrando a expressão da UCP2 em ambos os grupos são apresentados na Figura 1A. A expressão relativa da UCP2 foi 16% menor no grupo hipóxia do que no grupo controle, (0,82 [0,71-0,94] vs. 0,98 [0,78-1,19]), embora a diferença entre os dois grupos não tenha sido significativa (p = 0.11).
Os níveis de glicemia de jejum (Figura 1B) foram 12% menores no grupo hipóxia do que no grupo controle (144 [115-156] vs. 163 [133-184] mg/dL; p = 0,025). Os níveis de insulina sérica de jejum (Figura 1C) foram 5% maiores no grupo hipóxia em comparação ao grupo controle (0,44 [0,42-0,46] vs. 0,42 [0,40-0,44] ng/mL; p = 0,01). Os níveis de glucagon sérico de jejum (Figura 1D) foram 37% menores no grupo hipóxia do que no grupo controle (6,84 [3,64-9,90] vs. 10,87 [7,43-14,21] ng/mL; p = 0,023).
Após a exposição à hipóxia intermitente, o valor médio do HOMA-IR (Figura 1E) foi 15% menor no grupo hipóxia do que no grupo controle, embora a diferença não tenha sido significativa (3,45 [3,00-4,17] vs. 4.06 [3,35-4,55]; p = 0,095). O valor médio do HOMA-β (Figura 1F) foi 21% maior no grupo hipóxia do que no grupo controle (46,6 [42-74] vs. 36,6 [29-52]; p = 0,014).
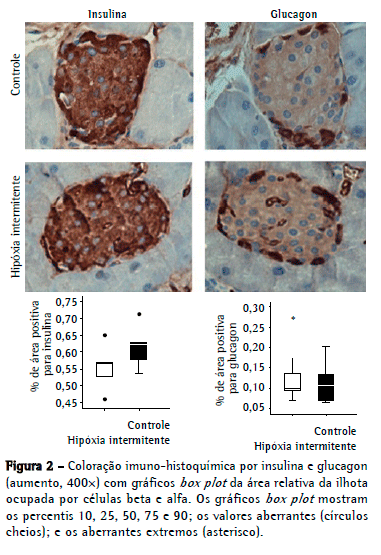
Conforme mostrado na Figura 2, a área relativa de células positivas para insulina no grupo hipóxia não foi significativamente maior do que no grupo controle: 0,62 [0,56-0,67] vs. 0,56 [0,51-0,59] respectivamente (p = 0,14). A área de células positivas para glucagon foi de 0,10 [0,06-0,15] no grupo hipóxia e de 0,09 [0,08-0,17] no grupo controle (p = 0,67).
Os parâmetros do perfil lipídico foram comparados entre os grupos. Os camundongos expostos a hipóxia intermitente não diferiram significativamente dos camundongos controle em relação aos triglicérides séricos de jejum e aos níveis de HDL, LDL e colesterol total.
Os pesos corporais permaneceram relativamente estáveis durante o período de estudo de 35 dias: 23,7 ± 0,3 g no dia 1 versus 25,1 ± 0,3 g no dia 35 no grupo controle; e 24,6 ± 0,5 g no dia 1 versus 24,8 ± 0,3 g no dia 35 no grupo hipóxia. O peso corporal inserido como variável dependente no modelo de EEG não mostrou nenhum efeito da alocação dos grupos ou da ingestão de alimentos. O tempo teve um efeito significativo, e houve uma interação significativa entre tempo e grupo (p < 0,001 para ambos).
DiscussãoSegundo nosso conhecimento, este foi o primeiro estudo a analisar o efeito de um modelo de AOS baseado em hipóxia intermitente sobre a expressão do mRNA da UCP2 no pâncreas. Nesse modelo, a exposição a 480 ciclos por dia de hipóxia intermitente com um nadir de FiO2 de 8% por 35 dias resultou em níveis de expressão do mRNA da UCP2 menores do que os observados para a condição controle, embora a diferença não tenha sido estatisticamente significativa. Esses resultados preliminares suplementam o conhecimento atual sobre os mecanismos subjacentes às consequências metabólicas da AOS, gerando diversas hipóteses. A ausência de redução significativa na expressão da UCP2 no grupo hipóxia pode indicar que a hipóxia intermitente não tem efeito sobre a expressão da UCP2 ou que ela promove a downregulation desta expressão para um grau abaixo do limiar do poder estatístico permitido pelo pequeno número de animais avaliados.
Os resultados do presente estudo contribuem para o debate sobre as alterações metabólicas induzidas pela AOS. Nossos dados mostram que os camundongos expostos a hipóxia intermitente apresentaram menores níveis séricos de glicose e glucagon de jejum do que os camundongos controle. Estudos anteriores demonstraram que camundongos expostos a hipóxia intermitente apresentam níveis de glicose elevados(10,21) ou diminuídos.(11) Nossos achados estão de acordo com os deste último estudo, que envolveu camundongos C57BL/6J magros e obesos expostos a hipóxia intermitente crônica por 5 dias.(11) Nos camundongos magros, os níveis de glicose e a resistência à insulina diminuíram após hipóxia. Nos camundongos obesos (deficientes em leptina), os níveis de glicose também diminuíram, embora a resistência à insulina tenha aumentado.(11) No presente estudo, foram empregados apenas camundongos magros, e nossos resultados podem, portanto, representar a resposta real de camundongos magros expostos a hipóxia intermitente, evidenciada pelo aumento dos níveis de insulina e diminuição dos níveis de glucagon.
Nosso achado de que os níveis de insulina sérica de jejum e a função das células beta foram maiores no grupo hipóxia do que no grupo controle é compatível com os níveis menores de glicose e glucagon relatados anteriormente em resposta à hipóxia. Em estudos anteriores, os camundongos obesos expostos a hipóxia intermitente apresentaram maiores níveis de insulina do que os camundongos controle,(11,21,22) embora a FiO2 e o período de exposição tenham diferido dos empregados no presente estudo. Outro estudo mostrou que os níveis de insulina foram menores nos camundongos expostos a hipóxia intermitente do que nos camundongos controle.(23)
No presente estudo, os níveis de insulina foram maiores nos camundongos expostos a hipóxia intermitente do que nos camundongos controle. Os níveis significativamente menores de glucagon encontrados no grupo hipóxia podem indicar que mecanismos compensatórios são ativados para manter a homeostase da glicose. Uma revisão da literatura não encontrou estudos comparáveis que descrevam a resposta do glucagon à hipóxia intermitente.
Nenhuma diferença na resistência à insulina foi detectada pelo HOMA-IR nos camundongos submetidos a hipóxia intermitente, sendo que os níveis de glicose dos mesmos foram maiores do que os dos controles. O valor do HOMA-β foi maior no primeiro grupo, sugerindo que algum mecanismo - talvez uma redução dos níveis de UCP2 - foi ativado para aumentar a produção de insulina. O HOMA-IR e o HOMA-β não foram validados para uso em roedores, sendo que a fórmula para este último representa uma medida da função das células beta em humanos.(24) Porém, a diferença na relação insulina/glicose sugere que os camundongos podem se tornar resistentes à insulina, como relatado anteriormente.(11)
Até o momento, são poucos os relatos na literatura que abordam a função das células beta pancreáticas e o sono. No experimento aqui relatado, os valores do HOMA-β foram significativamente elevados nos animais expostos a hipóxia intermitente por 35 dias, sendo 10% maiores no grupo hipóxia do que no grupo controle. O aumento no valor do HOMA-β indica que a AOS causa um aumento excessivo da demanda funcional das células beta pancreáticas, o que leva á piora da secreção de insulina com o avançar da doença.
Nossos achados devem ser interpretados com cautela. Os dados referentes aos níveis de glicose podem não ser confiáveis, pois mesmo os camundongos do grupo controle tinham níveis altos de glicose, apesar do jejum. Uma possível explicação para isso, além de um erro de medição, é que, embora a ração padrão tenha sido removida das gaiolas, uma porção significativa poderia ter permanecido dentro da gaiola. Porém, o erro pode ter sido sistemático, sendo o menor nível de glicose no grupo hipóxia um efeito do maior nível de insulina. Embora o resultado seja negativo, futuras explorações sobre o efeito da hipóxia intermitente sobre a UCP2 poderiam ser úteis. Modelos empregando camundongos knockout para a UCP2 poderiam esclarecer a verdadeira reação dessa proteína à hipóxia intermitente. Um teste de provocação oral de glicose poderia confirmar a resistência à insulina, e uma curva seria mais informativa do que medições individuais. Embora difícil em camundongos, isso foi feito anteriormente.(6) Os triglicérides baixos sugerem que os camundongos do grupo hipóxia não estavam comendo tanto quanto os do grupo controle. Isso poderia ser um potencial fator de confusão dos achados.
A hiperlipidemia é comum em pessoas com apneia do sono. Experimentos avaliando o efeito da hipóxia intermitente sobre o perfil lipídico relataram achados discordantes em razão de inconsistências metodológicas entre os estudos. Exemplos de parâmetros que variam de estudo para estudo e poderiam explicar achados discrepantes são diferenças na duração da exposição (em dias e em horas por dia), na duração de cada ciclo de hipóxia (em segundos), nos nadires de FiO2 e nas linhagens de camundongos. A magnitude do distúrbio metabólico induzido pela hipóxia intermitente depende do nadir de FiO2. Experimentos utilizando FiO2 de 10% como nadir do ciclo de hipóxia não induziram alterações metabólicas após 28 dias. Por outro lado, a utilização de um nadir de FiO2 de 5% levou a alterações em todos os parâmetros do perfil lipídico.(25) A gravidade moderada da hipóxia intermitente - tanto em termos do nadir de FiO2 quanto das horas de exposição por dia - pode ser um motivo para a ausência de alterações nos parâmetros lipídicos no presente estudo. Da mesma forma, o nadir de FiO2 de 8% e as 8 h de exposição por dia aqui empregadas podem não ter induzido uma hipóxia grave o suficiente para afetar a expressão da UCP2.
Em suma, nossa investigação sobre o efeito da hipóxia intermitente sobre a expressão do mRNA da UCP2 em camundongos C57BL revelou que, após 35 dias de exposição a hipóxia intermitente, não houve alterações significativas na expressão dessa proteína regulatória. No nível de gravidade aqui empregado, a hipóxia intermitente causa alterações na função pancreática que podem não estar relacionadas a potenciais alterações na expressão do mRNA da UCP2.
AgradecimentosOs autores gostariam de agradecer à Professora Doutora Luise Meurer seu papel determinante na análise imuno-histoquímica.
Referências1. Dempsey JA, Veasey SC, Morgan BJ, O'Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90(1): 47-112. http://dx.doi.org/10.1152/physrev.00043.2008
2. Tufik S, Santos-Silva R, Taddei JA, Bittencourt LR. Obstructive sleep apnea syndrome in the Sao Paulo Epidemiologic Sleep Study. Sleep Med. 2010;11(5):441-6. http://dx.doi.org/10.1016/j.sleep.2009.10.005
3. Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62(7):569-76. http://dx.doi.org/10.1016/j.jacc.2013.05.045
4. Trombetta IC, Somers VK, Maki-Nunes C, Drager LF, Toschi-Dias E, Alves MJ, et al. Consequences of comorbid sleep apnea in the metabolic syndrome - implications for cardiovascular risk. Sleep. 2010;33(9):1193-9.
5. Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A, et al. Sleep apnea and cardiovascular disease: an American Heart Association/American College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation. 2008;118(10):1080-1111. http://dx.doi.org/10.1161/CIRCULATIONAHA.107.189375
6. Yokoe T, Alonso LC, Romano LC, Rosa TC, O'Doherty RM, Garcia-Ocana A, et al. Intermittent hypoxia reverses the diurnal glucose rhythm and causes pancreatic beta-cell replication in mice. J Physiol. 2008;586(3):899-911. http://dx.doi.org/10.1113/jphysiol.2007.143586
7. Morgenstern M, Wang J, Beatty N, Batemarco T, Sica AL, Greenberg H. Obstructive sleep apnea: an unexpected cause of insulin resistance and diabetes. Endocrinol Metab Clin North Am. 2014;43(1):187-204. http://dx.doi.org/10.1016/j.ecl.2013.09.002
8. Weinstock TG, Wang X, Rueschman M, Ismail-Beigi F, Aylor J, Babineau DC, et al. A controlled trial of CPAP therapy on metabolic control in individuals with impaired glucose tolerance and sleep apnea. Sleep. 2012;35(5):617-25B. http://dx.doi.org/10.5665/sleep.1816
9. Dorkova Z, Petrasova D, Molcanyiova A, Popovnakova M, Tkacova R. Effects of continuous positive airway pressure on cardiovascular risk profile in patients with severe obstructive sleep apnea and metabolic syndrome. Chest. 2008;134(4):686-92. http://dx.doi.org/10.1378/chest.08-0556
10. Iiyori N, Alonso LC, Li J, Sanders MH, Garcia-Ocana A, O'Doherty RM, et al. Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am J Respir Crit Care Med. 2007;175(8):851-7. http://dx.doi.org/10.1164/rccm.200610-1527OC
11. Polotsky VY, Li J, Punjabi NM, Rubin AE, Smith PL, Schwartz AR, et al. Intermittent hypoxia increases insulin resistance in genetically obese mice. J Physiol. 2003;552(Pt 1):253-64. http://dx.doi.org/10.1113/jphysiol.2003.048173
12. Liu J, Li J, Li WJ, Wang CM. The role of uncoupling proteins in diabetes mellitus. J Diabetes Res. 2013;2013:585897. http://dx.doi.org/10.1155/2013/585897
13. Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, et al. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105(6):745-55. http://dx.doi.org/10.1016/S0092-8674(01)00378-6
14. Chan CB, De Leo D, Joseph JW, McQuaid TS, Ha XF, Xu F, et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. 2001;50(6):1302-10. http://dx.doi.org/10.2337/diabetes.50.6.1302
15. Echtay KS. Mitochondrial uncoupling proteins--what is their physiological role? Free Radic Biol Med. 2007;43(10):1351-71. http://dx.doi.org/10.1016/j.freeradbiomed.2007.08.011
16. Pi J, Collins S. Reactive oxygen species and uncoupling protein 2 in pancreatic -cell function. Diabetes Obes Metab. 2010; Suppl 2:141-8. http://dx.doi.org/10.1111/j.1463-1326.2010.01269.x
17. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press (US), 2011.
18. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402-8. http://dx.doi.org/10.1006/meth.2001.1262
19. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499-502.
20. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412-9. http://dx.doi.org/10.1007/BF00280883
21. Reinke C, Bevans-Fonti S, Drager LF, Shin MK, Polotsky VY. Effects of different acute hypoxic regimens on tissue oxygen profiles and metabolic outcomes. J Appl Physiol (1985). 2011;111(3):881-90. http://dx.doi.org/10.1152/japplphysiol.00492.2011
22. Drager LF, Li J, Reinke C, Bevans-Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity (Silver Spring). 2011;19(11):2167-74. http://dx.doi.org/10.1038/oby.2011.240
23. Savransky V, Nanayakkara A, Vivero A, Li J, Bevans S, Smith PL, et al. Chronic intermittent hypoxia predisposes to liver injury. Hepatology. 2007;45(4):1007-13. http://dx.doi.org/10.1002/hep.21593
24. Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27(6):1487-95. http://dx.doi.org/10.2337/diacare.27.6.1487
25. Li J, Savransky V, Nanayakkara A, Smith PL, O'Donnell CP, Polotsky VY. Hyperlipidemia and lipid peroxidation are dependent on the severity of chronic intermittent hypoxia. J Appl Physiol (1985). 2007;102(2):557-63. http://dx.doi.org/10.1152/japplphysiol.01081.2006
*Trabalho realizado no Programa de Pós-Graduação em Medicina: Ciências Médicas, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Endereço para correspondência: Carolina Caruccio Montanari. Rua Ramiro Barcelos, 2350, CEP 90035-903, Porto Alegre, RS, Brasil.
Tel. 55 51 3359-8289. E-mail: carolmontanari@gmail.com
Apoio financeiro: Este estudo recebeu apoio financeiro do Fundo de Incentivo à Pesquisa do Hospital de Clínicas de Porto Alegre (FIPE-HCPA) e do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; bolsa de doutorado para CZ Fiori e bolsa de pesquisa para D Martinez).
Recebido para publicação em 17/9/2014. Aprovado, após revisão, em 5/12/2014.
Sobre os autoresLuciana Rodrigues Vieira
Mestranda em Ciências Médicas. Programa de Pós-Graduação em Medicina: Ciências Médicas, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Denis Martinez
Professor Associado III. Faculdade de Medicina, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Luiz Felipe Forgiarini
Professor. Faculdade Anhanguera de Pelotas, Pelotas (RS) Brasil.
Darlan Pase da Rosa
Professor. Faculdade Cenecista de Bento Gonçalves, Bento Gonçalves; e Professor Colaborador, Universidade Luterana do Brasil, Canoas (RS) Brasil.
Gustavo Alfredo Ochs de Muñoz
Mestrando. Programa de Pós-Graduação em Saúde da Criança e do Adolescente, Universidade do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Micheli Fagundes
Mestranda. Programa de Pós-Graduação em Ciências da Saúde: Cardiologia e Ciências Cardiovasculares, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Emerson Ferreira Martins
Mestrando. Programa de Pós-Graduação em Ciências da Saúde: Cardiologia e Ciências Cardiovasculares, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Carolina Caruccio Montanari
Mestranda. Programa de Pós-Graduação em Medicina: Ciências Médicas, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.
Cintia Zappe Fiori
Doutoranda. Programa de Pós-Graduação em Ciências da Saúde: Cardiologia e Ciências Cardiovasculares, Universidade Federal do Rio Grande do Sul, Porto Alegre (RS) Brasil.












 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket