
Continuous and bimonthly publication
ISSN (on-line): 1806-3756



Elisangela Jacinto de Faria, Isabel Cristina Jacinto de Faria, José Dirceu Ribeiro, Antônio Fernando Ribeiro, Gabriel Hessel, Carmen Sílvia Bertuzzo
ABSTRACT
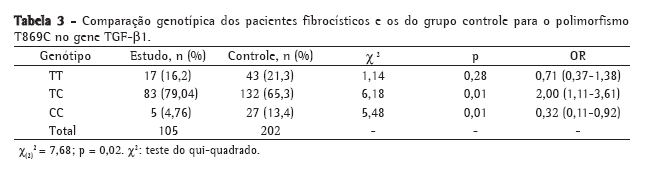
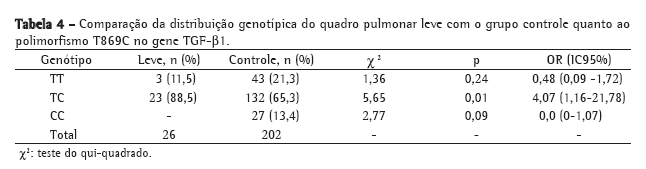
Objective: To identify associations between genetic polymorphisms (in the MBL2, TGF-β1 and CD14 genes) and the severity of the lung disease in patients with cystic fibrosis (CF), as well as between the presence of ΔF508 alleles and lung disease severity in such patients. Methods: This was a cross-sectional cohort study, based on clinical and laboratory data, involving 105 patients with CF treated at a university hospital in the 2005-2006 period. We included 202 healthy blood donors as controls for the determination of TGF-β1 and CD14 gene polymorphisms. Polymorphisms in the MBL2 and TGF-β1 genes at codon 10, position +869, were genotyped using the allele-specific PCR technique. The C-159T polymorphism in the CD14 gene was genotyped using PCR and enzymatic digestion. Results: Of the 105 CF patients evaluated, 67 presented with severe lung disease according to the Shwachman score. The MBL2 gene polymorphisms were not associated with disease severity in the CF patients. Analysis of the T869C polymorphism in the TGF-β1 gene showed an association only between TC heterozygotes and mild pulmonary disease. Although patients presenting the TT genotype of the C159T polymorphism in the CD14 gene predominated, there was no significant difference regarding lung disease severity. Conclusions: There was an association between the TC genotype of the T869C polymorphism (TGF-β1) and mild pulmonary disease in CF patients. In the CD14 gene, the TT genotype seems to be a risk factor for pulmonary disease but is not a modulator of severity. We found no association between being a ΔF508 homozygote and presenting severe lung disease.
Keywords: Cystic fibrosis; Polymorphism, genetic; Severity of illness index; Mannose-binding lectin; Transforming growth factor beta.
RESUMO
Objetivo: Verificar a correlação entre os polimorfismos dos genes MBL2, TGF-β1 e CD14 com a gravidade da doença pulmonar em pacientes com fibrose cística (FC), bem como correlacionar a presença dos alelos ΔF508 com a gravidade da doença naqueles pacientes. Métodos: Estudo clínico-laboratorial, de corte transversal, com 105 pacientes fibrocísticos de um hospital universitário em 2005-2006. Foram analisados 202 doadores de sangue saudáveis como controles para a pesquisa dos polimorfismos no gene TGF-β1 e CD14. A análise de polimorfismos nos genes MBL2 e TGF-β1 no códon 10, posição +869, foi realizada pela técnica da PCR alelo-específica. A genotipagem do polimorfismo C-159T no gene CD14 foi realizada através de PCR e digestão enzimática. Resultados: Dos 105 pacientes com FC avaliados, 67 apresentavam doença pulmonar grave segundo o escore de Shwachman. Os polimorfismos do gene MBL2 não foram associados com a gravidade da doença nos fibrocísticos. A análise do polimorfismo T869C no gene TGF-β1 mostrou somente uma associação entre o heterozigoto TC com doença pulmonar leve. Para o polimorfismo C-159T no gene CD14, obtivemos um predomínio de pacientes com o genótipo TT, mas não houve diferença significativa com relação à gravidade do quadro pulmonar. Conclusões: Houve associação entre o genótipo TC do polimorfismo T869C (TGF-β1) e o quadro pulmonar leve nos fibrocísticos. No gene CD14, o genótipo TT parece ser um fator de risco para o quadro pulmonar, mas não um fator modulador da gravidade. Não existiu associação entre pacientes homozigotos para a mutação ΔF508 e a gravidade do quadro pulmonar.
Palavras-chave: Fibrose cística; Polimorfismo genético; Índice de gravidade de doença; Lectina de ligação a manose; Fator transformador de crescimento beta.
Introdução









Sociedade Brasileira de Pneumologia e Tisiologia
Secretaria do Jornal Brasileiro de Pneumologia
SCS Quadra 01, Bloco K, Salas 203/204 Ed. Denasa
Brasília - DF CEP: 70.398-900
Fone/fax: 08000 61 6218, (61) 3245 1030 e (61) 3245 6218
E-mail: jbp@sbpt.org.br
Development by: ![]()
© All rights reserved 2024 - Jornal Brasileiro de Pneumologia

 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket