ABSTRACT
Objective: To study the expression of COX-1 and COX-2 in the remodeled lung in systemic sclerosis (SSc) and idiopathic pulmonary fibrosis (IPF) patients, correlating that expression with patient survival. Methods: We examined open lung biopsy specimens from 24 SSc patients and 30 IPF patients, using normal lung tissue as a control. The histological patterns included fibrotic nonspecific interstitial pneumonia (NSIP) in SSc patients and usual interstitial pneumonia (UIP) in IPF patients. We used immunohistochemistry and histomorphometry to evaluate the expression of COX-1 and COX-2 in alveolar septa, vessels, and bronchioles. We then correlated that expression with pulmonary function test results and evaluated its impact on patient survival. Results: The expression of COX-1 and COX-2 in alveolar septa was significantly higher in IPF-UIP and SSc-NSIP lung tissue than in the control tissue. No difference was found between IPF-UIP and SSc-NSIP tissue regarding COX-1 and COX-2 expression. Multivariate analysis based on the Cox regression model showed that the factors associated with a low risk of death were younger age, high DLCO/alveolar volume, IPF, and high COX-1 expression in alveolar septa, whereas those associated with a high risk of death were advanced age, low DLCO/alveolar volume, SSc (with NSIP), and low COX-1 expression in alveolar septa. Conclusions: Our findings suggest that strategies aimed at preventing low COX-1 synthesis will have a greater impact on SSc, whereas those aimed at preventing high COX-2 synthesis will have a greater impact on IPF. However, prospective randomized clinical trials are needed in order to confirm that.
Keywords:
Scleroderma, systemic; Idiopathic pulmonary fibrosis; Inflammation; Survival rate.
RESUMO
Objetivo: Estudar a expressão de COX-1 e COX-2 em áreas pulmonares remodeladas em pacientes com esclerose sistêmica (ES) ou fibrose pulmonar idiopática (FPI) e correlacioná-la com a sobrevida desses pacientes. Métodos: Examinamos espécimes de biópsia pulmonar a céu aberto de 24 pacientes com ES e de 30 pacientes com FPI, utilizando-se tecido pulmonar normal como controle. Os padrões histológicos incluíram pneumonia intersticial não específica (PINE) fibrótica em pacientes com ES e pneumonia intersticial usual (PIU) nos pacientes com FPI. Imuno-histoquímica e histomorfometria foram usadas para avaliar a expressão celular de COX-1 e COX-2 em septos alveolares, vasos e bronquíolos, sua correlação com provas de função pulmonar e seu impacto na sobrevida. Resultados: A expressão de COX-1 e COX-2 em septos alveolares foi significativamente maior em FPI-PIU e ES-PINE do que no tecido controle. Não houve diferença entre FPI-PIU e ES-PINE quanto à expressão de COX-1 e COX-2. A análise multivariada baseada no modelo de regressão de Cox mostrou que os fatores associados a baixo risco de morte foram ter idade menor, valores elevados de DLCO/volume alveolar, FPI, e alta expressão de COX-1 em septos alveolares, ao passo que os fatores associados a alto risco de morte foram ter idade maior, valores baixos de DLCO/volume alveolar, ES (com PINE) e baixa expressão de COX-1 em septos alveolares. Conclusões: Nossos resultados sugerem que estratégias de prevenção de baixa síntese de COX-1 terão maior impacto sobre a ES, ao passo que as de prevenção de alta síntese de COX-2 terão maior impacto sobre a FPI. Porém, são necessários ensaios clínicos randomizados prospectivos para confirmar essa hipótese.
Palavras-chave:
Escleroderma sistêmico; Fibrose pulmonar idiopática; Inflamação; Taxa de sobrevida.
IntroductionLung remodeling is a common end-stage sequela of idiopathic pulmonary fibrosis (IPF) and systemic sclerosis (SSc), resulting in disruption of lung architecture, leading to progressive respiratory failure.(1-4) Histologically, the remodeling process is characterized by diffuse chronic interstitial inflammation and increased fibroblast proliferation, as well as by increased extracellular matrix synthesis and collagen deposition.(2,5,6) Therefore, modulation of inflammation, fibroblast proliferation, and collagen synthesis by effector mediators in IPF and SSc is very important. Despite the characterization of a variety of key participants, the mediators and mechanisms involved in the pathogenesis of IPF and SSc have yet to be fully defined, which might explain the limited number of therapeutic approaches, with little impact on long-term survival.(7,8)
It is known that COX is the key enzyme in the conversion of arachidonic acid to prostaglandin E2 (PGE2), the precursor of a diverse family of bioactive lipid mediators including prostaglandins, thromboxane, and prostacyclin. It exists in two isoforms, namely COX-1 and COX-2. The former is constitutively expressed in most tissues and acts as a housekeeping enzyme regulating vascular homeostasis, protecting the gastric mucosa, and maintaining renal integrity,(9,10) whereas the latter has lower levels of expression in most tissues but is inducible in response to growth factors, cytokines, and other proinflammatory molecules.(11-13)
Regarding the proinflammatory and anti-inflammatory roles of COX-1 and COX-2, immunohistochemistry can be a useful tool to detect COX-1 and COX-2 in the remodeled lung in patients with SSc and IPF. Data on the assessment of COX-1 and COX-2 in the remodeled lung have previously been reported in serum(14,15) and bronchoalveolar lavage fluid(16) from patients with SSc, as well as in fibroblast cultures(4) and biopsies(17,18) from patients with IPF. However, the roles of COX-1 and COX-2 in the mechanisms involved in the remodeled lung in IPF and SSc patients are still unclear, and there has been uncertainty regarding the best way to detect COX-2. The aim of the present study was to study the expression of COX-1 and COX-2 in lung biopsy specimens (COX-1 and COX-2 expression being separately evaluated in alveolar septa, bronchioles, and vessels) and correlate it with patient survival.
MethodsBetween January of 2002 and July of 2008, 24 consecutive patients with SSc and interstitial lung disease and 30 patients suspected of having IPF on the basis of HRCT findings were submitted to open lung biopsy at the University of São Paulo School of Medicine Hospital das Clínicas, located in the city of São Paulo, Brazil. All patients fulfilled the diagnostic criteria for SSc(19) and IPF(1) Open lung biopsy was performed by formal thoracotomy, areas of honeycombing being avoided. All patients gave written informed consent, and the study was approved by the local research ethics committee (Protocol no. 0960/08).
We analyzed the clinical records of all patients. Disease duration was determined on the basis of the onset of the first symptom. Pulmonary function testing and HRCT were performed within up to 3 months before the biopsy. Pulmonary function testing included VC, FEV1, FVC, FEV1/FVC, TLC, RV, and DLCO. Physiological assessment was performed before open lung biopsy and before the initiation of treatment. All pulmonary function tests, including spirometry, determination of lung volumes, and measurement of DLCO, were performed on the same day. All spirometric tests were performed with a calibrated pneumotachograph (Medical Graphics Co., St. Paul, MN, USA), all values being expressed as a percentage of their respective predicted value, the reference values having been established by Pereira et al.(20) Lung volumes were measured with a whole-body plethysmograph (Medical Graphics Co.), all values being expressed as a percentage of the predicted values.(21) Diffusing capacity was expressed as a percentage of the predicted values.(22) Diffusing capacity was expressed as a percentage of the predicted values.(23) All patients were followed regularly after treatment until death, blood and lung function tests being regularly performed. The primary endpoint was to evaluate the impact of COX-1 and COX-2 changes on survival and analyze differences between SSc and IPF. Table 1 shows the demographic data. As a control, normal lung tissue was obtained from 10 individuals (6 males and 4 females) whose median age was 46.6 5.8 years and who had died suddenly of nonpulmonary causes.
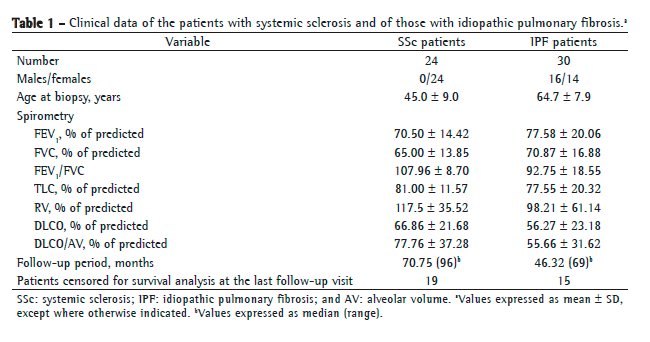
Regarding open lung biopsy findings, usual interstitial pneumonia (UIP), the histological pattern of IPF, was characterized by patchy subpleural and paraseptal distribution of parenchymal injury. Temporal heterogeneity was seen at low magnification, areas of normal lung parenchyma alternating with alveolar collapse, interstitial mononuclear infiltrates, septal fibromyxoid tissue (fibroblastic foci), and honeycomb lung.(2) All of the patients with SSc had histological patterns consistent with fibrotic nonspecific interstitial pneumonia (NSIP), as defined by temporally homogeneous septal thickening and interstitial fibrosis.(19)
For immunohistochemistry analysis, a standard peroxidase technique was used (Harris's hematoxylin being used as the counterstain) in order to identify COX-1 and COX-2 expression in alveolar septa, bronchiolar walls, and vascular walls in normal lung tissue (the control tissue), in lung tissue showing the UIP pattern (the UIP tissue), and in lung tissue showing the NSIP pattern (the NSIP tissue). All antibodies used were biotinylated goat polyclonal antibodies. Anti-COX-1 and anti-COX-2 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were incubated with tissue sections at dilutions of 1:50 and 1:100, respectively. The Novolink Max Polymer amplification kit (Leica Biosystems Newcastle Ltd, Newcastle upon Tyne, UK) was used for signal amplification, and 3,3'-diaminobenzidine tetrahydrochloride (0.25 mg dissolved in 1 mL of 0.02% hydrogen peroxide) was used as a precipitating substrate for signal detection. The specificity of primary antibodies was confirmed by appropriate reagent controls (the primary antibody being omitted or nonimmune serum being substituted for the primary antibody in the staining protocol), which revealed no staining.
Regarding histomorphometry, COX-1 expression and COX-2 expression were assessed by a point-counting technique in 50 and 30 fields in alveolar septa, bronchiolar walls, and vascular walls in the control tissue, in the UIP tissue, and in the NSIP tissue. The technique was performed with a 100-point grid (area, 187,500 m(21); magnification, ×400) attached to the microscope eyepiece.(23) At a magnification of ×400, the septal, bronchiolar, and vascular areas in each field were calculated on the basis of the number of points overlying connective tissue, as a proportion of the total grid area. Subsequently, the number of immunostained cells within the septal, bronchiolar, and vascular areas was counted. The areal fraction of immunostained cells represents the percentage ratio of the area of labeled cells in relationship to the total area covered by the grid in the eyepiece.
In order to assess interobserver variability, we compared the results obtained by two observers in 20% of the slides. The coefficient of variation for the interobserver error of the cell count was 5%.
Data are presented as mean SD and 95% CI. The Student's t-test for independent samples was used in order to test the relationship between continuous variables, and the residuals were examined to ensure that they were approximately normally distributed. The relationship between cellularity (as determined by immunostaining) and pulmonary function test results was evaluated by Pearson's correlation coefficient. For all cases, measured variable values were arranged in ascending order and divided into two groups on the basis of the median value of each variable. For each variable, the groups were designated low degree and high degree, as follows: alveolar septal COX-1 (low degree, < 2.35%; high degree, 2.35%); vascular COX-1 (low degree, < 2.91%; high degree, 2.91%); bronchiolar COX-1 (low degree, < 2.88%; high degree, 2.88%); total COX-1 (low degree, < 2.77%; high degree, 2.77%); alveolar septal COX-2 (low degree, < 2.04%; high degree, 2.04%); vascular COX-2 (low degree, < 2.34%; high degree, 2.34%); bronchiolar COX-2 (low degree, < 2.34%; high degree, 2.34%); and total COX-2 (low degree, < 2.16%; high degree, 2.16%).
Overall survival analysis was performed in two steps. First, we performed a univariate analysis relating overall follow-up to each of the measured variables by means of the Kaplan-Meier method and then analyzed survival using the log-rank test. The variables that were found to be significant in the univariate analysis were included in the multivariate analysis based on the Cox proportional hazards regression model. A positive event was defined as any death caused by IPF or SSc. Deaths from causes other than IPF or SSc and living patients were included in the models as censored cases.
All statistical procedures were performed with the Statistical Package for the Social Sciences, version 18.0 (SPSS Inc., Chicago, IL, USA). For all tests, the significance level was set at 5%.
Results
Table 1 summarizes the clinical features of the patients with SSc (n = 24) and those of those with IPF (n = 30). Six of 17 SSc patients (35.29%) and 13 of 19 IPF patients (68.42%) had restrictive lung disease. Respiratory function test results were as follows: FVC < 80% in 18 (75%) of the 24 SSc patients and in 19 of 22 IPF patients (86.36%); TLC < 80% in 6 of 17 SSc patients (35.9%) and in 13 of 19 IPF patients (68.42%); DLCO < 80% in 12 of 15 SSc patients (80%) and in 8 of 9 IPF patients (88.88%); and DLCO/alveolar volume < 80% in 11 of 18 SSc patients (61.11%) and in 11 of 14 IPF patients (78.57%). A significant negative correlation was found between COX-2 expression in vessels and FVC (r= −0.28; p = 0.05), as well as between COX-2 expression in alveolar septa and DLCO (r = −0.80; p = 0.009).
Figure 1 shows alveolar septa, vessels, and bronchioles in the control tissue, in the NSIP tissue, and in the UIP tissue immunostained for COX-1 (in A, C, E, G, I, K, M, O, and Q) and COX-2 (in B, D, F, H, J, L, N, P, and R). The NSIP and UIP tissues differed from the control tissue in terms of the immunostaining intensity of epithelial cells, endothelial cells, myofibroblasts, and smooth muscle cells in the alveolar septa, vessels, and bronchioles.
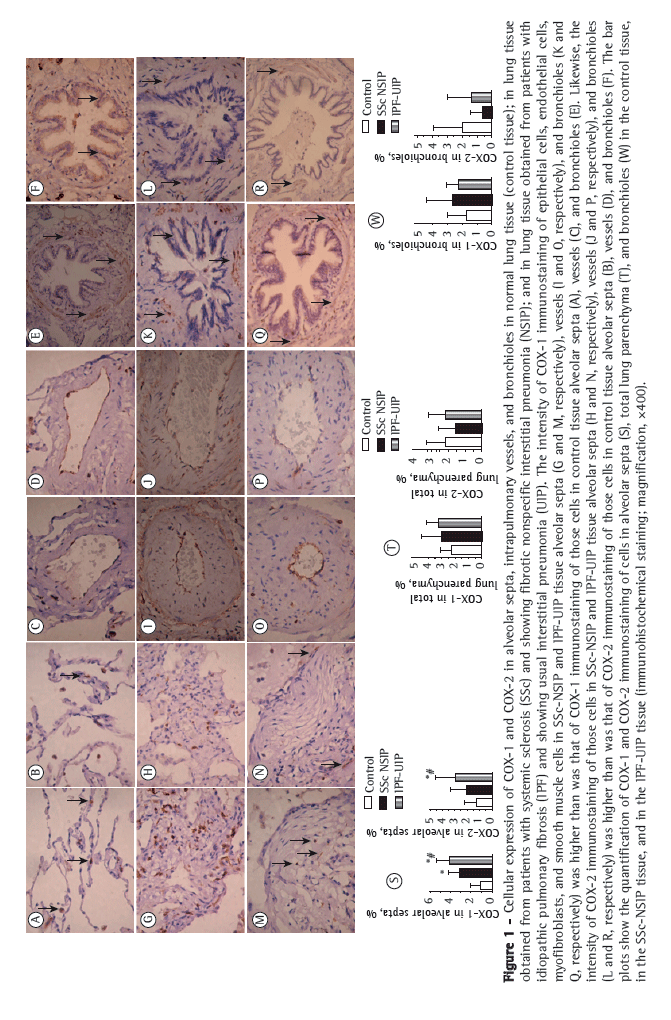
Table 2 summarizes the morphometric results. The proportion of alveolar septal cells immunostained for COX-1 and COX-2 was significantly higher in the UIP and NSIP tissues than in the control tissue. In other words, high proportions of alveolar septal cells staining for COX-1 and COX-2 were associated with the UIP and NSIP patterns. As can be seen in the bar plots in Figure 1 (S and T) the relationship of COX-1 and COX-2 with IPF (the UIP pattern) was stronger than was that of COX-1 and COX-2 with SSc (the NSIP pattern). Although the proportion of bronchiolar cells immunostained for COX-2 was lower in the NSIP and UIP tissues than in the control tissue (Figure 1W), the difference was not statistically significant. In addition, although the proportion of bronchiolar cells immunostained for COX-1 was higher in the UIP and NSIP tissues than in the control tissue (Figure 1W), the difference was not significant. No differences were found among the tissues in terms of the COX-1 or COX-2 immunostaining, for vessels or for the total parenchyma (Table 2).
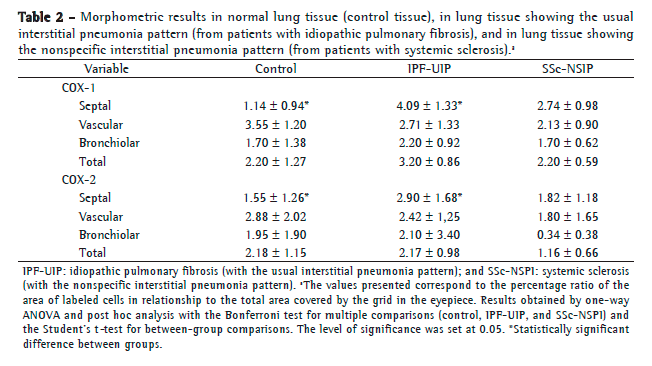
A preliminary analysis of the Kaplan-Meier survival curves showed that survival was better in the patients with SSc (the fibrotic NSIP pattern) and COX-2 expression > 2.25% (median survival, 70.75 months) than in those with IPF (the UIP pattern) and COX-2 expression < 2.25% (median survival, 46.32 months; Figure 2). Therefore, we coded the fibrotic NSIP pattern as a single dummy variable with a value of 1 and the UIP pattern with a value of 2. The results of the multivariate analysis based on the Cox proportional hazards regression model are shown in Table 3. After controlling for age, pulmonary function test results, the UIP pattern, and the fibrotic NSIP pattern, we found that only two variables were significantly associated with survival time: the fibrotic NSIP pattern and alveolar septal COX-2 (p = 0.02). Once these two variables were accounted for, none of the others were related to survival. The multivariate analysis showed a low risk of death for young patients with low FEV1/FVC, fibrotic NSIP pattern, and high-degree alveolar septal COX-2.
 Discussion
DiscussionThe limited number of therapeutic approaches that have any impact on long-term survival in patients with IPF-UIP and in those with SSc and fibrotic NSIP is due to the lack of definition regarding the mediators and mechanisms involved in the pathogenesis of IPF and SSc. Therefore, the question of interest is whether additional mediators can provide a better understanding of the pathogenesis of these diseases. The repair process involves two distinct stages: a regenerative, inflammatory phase, in which the microenvironment attempts to replace injured cells; and a fibrotic phase, in which connective tissue replaces normal parenchymal tissue.(24-26) In the repair process, PGE2 production by fibroblasts is increased,(27,28) which constitutes further evidence of the antiproliferative, anti-inflammatory and antifibrotic properties of COX-2/PGE2.(15) Therefore, our finding that immunohistochemistry staining for COX provides important information on the repair processes in pulmonary fibrosis is not surprising, and our results confirm that the expression of COX-2 is increased in IPF and SSc, with improved outcome in a group of patients. We found that the proportion of alveolar septal cells immunostained for COX-1 and COX-2 was significantly higher in lung tissue showing the UIP pattern and the fibrotic NSIP pattern than in normal lung tissue. Increased COX-1 expression was expected because COX-1 is constitutively expressed in most cells and tissues, whereas COX-2 is induced by inflammatory or mitogenic stimuli.(9) These results contrast with those of previous studies investigating IPF.(4,17,18) Those studies showed reduced COX-2 expression in pulmonary fibroblasts secondary to decreased COX-2 production. However, in those studies, COX-2 expression was measured only in fibroblasts, whereas in our study it was measured in the alveolar septa, including epithelial cells and fibroblasts in normal areas, collapsed areas, and fibroblast foci. Other studies, including a study by Lappi-Blanco et al.,(3) found increased expression of COX-2 in metaplastic epithelium and fibroblasts from fibrotic areas in IPF-UIP. These contradictory findings suggest that COX-2 plays a dual role in IPF-UIP. First, reduced COX-2 expression in normal areas, collapsed areas, and fibroblastic foci suggests an anti-inflammatory role for COX-2 in early-stage IPF-UIP. Second, the presence of progressive fibrosis even in the presence of increased COX-2 expression suggests that fibroblasts are unable to respond to stimulation by COX-2 and its main product (PGE2) so as to inhibit fibroblast proliferation, myofibroblastic transformation, and increased production of collagen and other extracellular matrix molecules.
In the present study, the proportion of alveolar septal cells immunostained for COX-1 and COX-2 was found to be lower in fibrotic NSIP tissue (from SSc patients) than in UIP tissue (from IPF patients). This finding contrasts with those of previous studies showing that COX-2 levels are higher in SSc patients.(14-16) In addition, COX-2 production has been shown to be much greater in the inflammatory resolution phase than in the early phase.(29) These contradictory findings suggest that COX-2 has a dual role in a normal inflammatory process, playing a proinflammatory role in the early phase and an anti-inflammatory role in the resolution phase.(29) Therefore, in view of the abovementioned evidence and of the latent inflammation in patients with SSc and lung involvement, our results emphasize the idea that COX-2 does not exert its anti-inflammatory effect properly, because there is inflammation even when COX-2 expression is increased in patients with SSc and fibrotic NSIP. However, further studies are needed in order to clarify the real reason why the COX-2 mechanism is deficient. We hypothesize that this is due to an inability of COX-2 to stimulate the production of PGE2 or other anti-inflammatory mediators in opposition to its own proinflammatory effects or an inability of the cells to respond appropriately to COX-2.
Our study has clinical and functional impact. We sought to establish a correlation between COX-2 and patient survival controlled for age, pulmonary function test results, the UIP pattern (in patients with IPF) and the NSIP pattern (in patients with SSc). Our multivariate analysis showed a low risk of death for younger patients with low DLCO/alveolar volume, SSc (and the NSIP histological pattern), high-degree total COX-2, and high-degree alveolar septal COX-1.
In conclusion, the expression of COX-1 and COX-2 in the lung parenchyma offers us the potential to control repair processes involved in the progression of SSc-NSIP and IPF-UIP, suggesting that strategies aimed at preventing low COX-1 synthesis will have a greater impact on SSc, whereas those aimed at preventing high COX-2 synthesis will have a greater impact on IPF. Prospective randomized trials are required in order to confirm that.
References1. Sociedade Brasileira de Pneumologia e Tisiologia. Diretrizes de Doenças Pulmonares Intersticiais da Sociedade Brasileira de Pneumologia e Tisiologia. J Bras Pneumol. 2012;38(Suppl 2):S1-S133.
2. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. American Thoracic Society; European Respiratory Society. Am J Respir Crit Care Med. 2002;165(2):277-304. Erratum in: Am J Respir Crit Care Med. 2002;166(3):426. http://dx.doi.org/10.1164/ajrccm.165.2.ats01 PMid:11790668
3. Lappi-Blanco E, Kaarteenaho-Wiik R, Maasilta PK, Anttila S, Pääkkö P, Wolff HJ. COX-2 is widely expressed in metaplastic epithelium in pulmonary fibrous disorders. Am J Clin Pathol. 2006;126(5):717-24. http://dx.doi.org/10.1309/PFGXCLNG2N17PJX9 PMid:17050069
4. Wilborn J, Crofford LJ, Burdick MD, Kunkel SL, Strieter RM, Peters-Golden M. Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesize prostaglandin E2 and to express cyclooxygenase-2. J Clin Invest. 1995;95(4):1861-8. http://dx.doi.org/10.1172/JCI117866 PMid:7706493 PMCid:PMC295728
5. Gabbiani G. The myofibroblast: a key cell for wound healing and fibrocontractive diseases. Prog Clin Biol Res. 1981;54:183-94. PMid:7015359
6. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345(7):517-25. http://dx.doi.org/10.1056/NEJMra003200 PMid:11519507
7. Dempsey OJ. Clinical review: idiopathic pulmonary fibrosis--past, present and future. Respir Med. 2006;100(11):1871-85. http://dx.doi.org/10.1016/j.rmed.2006.08.017 PMid:16987645
8. Downey DG, Brockbank S, Martin SL, Ennis M, Elborn JS. The effect of treatment of cystic fibrosis pulmonary exacerbations on airways and systemic inflammation. Pediatr Pulmonol. 2007;42(8):729-35. http://dx.doi.org/10.1002/ppul.20646 PMid:17588254
9. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12(12):1063-73. PMid:9737710
10. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97-120. http://dx.doi.org/10.1146/annurev.pharmtox.38.1.97 PMid:9597150
11. Xie WL, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc Natl Acad Sci U S A. 1991;88(7):2692-6. http://dx.doi.org/10.1073/pnas.88.7.2692 PMid:1849272 PMCid:PMC51304
12. O'Banion MK, Sadowski HB, Winn V, Young DA. A serum- and glucocorticoid-regulated 4-kilobase mRNA encodes a cyclooxygenase-related protein. J Biol Chem. 1991;266(34):23261-7. PMid:1744122
13. Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992;89(16):7384-8. http://dx.doi.org/10.1073/pnas.89.16.7384
14. Bassyouni IH, Talaat RM, Salem TA. Serum concentrations of cyclooxygenase-2 in patients with systemic sclerosis: association with lower frequency of pulmonary fibrosis. J Clin Immunol. 2012;32(1):124-30. http://dx.doi.org/10.1007/s10875-011-9601-z PMid:21979415
15. Xiao R, Kanekura T, Yoshida N, Higashi Y, Yan KL, Fukushige T, et al. 9-Cis-retinoic acid exhibits antifibrotic activity via the induction of cyclooxygenase-2 expression and prostaglandin E2 production in scleroderma fibroblasts. Clin Exp Dermatol. 2008;33(4):484-90. http://dx.doi.org/10.1111/j.1365-2230.2008.02727.x PMid:18462443
16. Kowal-Bielecka O, Kowal K, Distler O, Rojewska J, Bodzenta-Lukaszyk A, Michel BA, et al. Cyclooxygenase- and lipoxygenase-derived eicosanoids in bronchoalveolar lavage fluid from patients with scleroderma lung disease: an imbalance between proinflammatory and antiinflammatory lipid mediators. Arthritis Rheum. 2005;52(12):3783-91. http://dx.doi.org/10.1002/art.21432 PMid:16320329
17. Xaubet A, Roca-Ferrer J, Pujols L, Ramírez J, Mullol J, Marin-Arguedas A, et al. Cyclooxygenase-2 is up-regulated in lung parenchyma of chronic obstructive pulmonary disease and down-regulated in idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2004;21(1):35-42. PMid:15127973
18. Petkova DK, Clelland CA, Ronan JE, Lewis S, Knox AJ. Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology. 2003;43(4):381-6. http://dx.doi.org/10.1046/j.1365-2559.2003.01718.x PMid:14511257
19. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202-5. PMid:3361530
20. Pereira CA, Sato T, Rodrigues SC. New reference values for forced spirometry in white adults in Brazil. J Bras Pneumol. 2007;33(4):397-406. http://dx.doi.org/10.1590/S1806-37132007000400008 PMid:17982531
21. Neder JA, Andreoni S, Castelo-Filho A, Nery LE. Reference values for lung function tests. I. Static volumes. Braz J Med Biol Res. 1999;32(6):703-17. PMid:10412549
22. Andreoni S, Lerario MC, Nery LE. Reference values for lung function tests. II. Maximal respiratory pressures and voluntary ventilation. Braz J Med Biol Res. 1999;32(6):719-27. PMid:10412550
23. Hsia CC, Hyde DM, Ochs M, Weibel ER; ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181(4):394-418. http://dx.doi.org/10.1164/rccm.200809-1522ST PMid:20130146
24. Meneghin A, Hogaboam CM. Infectious disease, the innate immune response, and fibrosis. J Clin Invest. 2007;117(3):530-8. http://dx.doi.org/10.1172/JCI30595 PMid:17332880 PMCid:PMC1804377
25. Oikonomou N, Harokopos V, Zalevsky J, Valavanis C, Kotanidou A, Szymkowski DE, et al. Soluble TNF mediates the transition from pulmonary inflammation to fibrosis. PLoS One. 2006;1:e108. http://dx.doi.org/10.1371/journal.pone.0000108 PMid:17205112 PMCid:PMC1762410
26. Pardo A, Selman M. Molecular mechanisms of pulmonary fibrosis. Front Biosci. 2002;7:d1743-61.
27. Kobayashi T, Narumiya S. Function of prostanoid receptors: studies on knockout mice. Prostaglandins Other Lipid Mediat. 2002;68-69:557-73. http://dx.doi.org/10.1016/S0090-6980(02)00055-2
28. McCann MR, Monemdjou R, Ghassemi-Kakroodi P, Fahmi H, Perez G, Liu S, et al. mPGES-1 null mice are resistant to bleomycin-induced skin fibrosis. Arthritis Res Ther. 2011;13(1):R6. http://dx.doi.org/10.1186/ar3226 PMid:21266028 PMCid:PMC3546456
29. Gilroy DW, Colville-Nash PR, Willis D, Chivers J, Paul-Clark MJ, Willoughby DA. Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999;5(6):698-701. http://dx.doi.org/10.1038/9550 PMid:10371510
*Study carried out in the Laboratory of Histomorphometry and Pulmonary Genetics, Department of Pathology, University of São Paulo School of Medicine, São Paulo, Brazil.
Correspondence to: Vera Luiza Capelozzi or Edwin Roger Parra. Laboratório de Histomorfometria e Genética Pulmonar, Departamento de Patologia, Faculdade de Medicina da Universidade de São Paulo, Av. Dr. Arnaldo, 455, sala 1143, CEP 01246-903, São Paulo, SP, Brasil.
Tel. 55 11 3066-7427. Fax: 55 11 5096-0761. E-mail: vcapelozzi@lim05.fm.usp.br or erparra20003@yahoo.com.br
Financial support: This study received financial support from the Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, National Council for Scientific and Technological Development) and the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, São Paulo Research Foundation; Grant no. 2008/53022-3).
Submitted: 22 June 2013. Accepted, after review: 23 October 2013.
**A versão completa em português deste artigo está disponível em www.jornaldepneumologia.com.br
About the authorsEdwin Roger Parra
Postdoctoral Student. University of São Paulo School of Medicine, São Paulo, Brazil.
Flavia Lin
Medical Student. University of São Paulo School of Medicine, São Paulo, Brazil.
Vanessa Martins
Master's Student. University of São Paulo School of Medicine, São Paulo, Brazil.
Maristela Peres Rangel
Doctoral Student. University of São Paulo School of Medicine, São Paulo, Brazil.
Vera Luiza Capelozzi
Associate Professor. Department of Pathology, University of São Paulo School of Medicine, São Paulo, Brazil.












 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket