
Continuous and bimonthly publication
ISSN (on-line): 1806-3756



Ricardo de Amorim Correa, Marcus Vinicius Souza Couto Moreira, Jucielle Marcelina da Silva Saraiva, Eliane Viana Mancuzo, Luciana Cristina dos Santos Silva, José Roberto Lambertucci
ABSTRACT
Schistosomiasis mansoni is the third most prevalent endemic parasitic disease in the world. It is estimated that over 200 million people are infected with parasites belonging to one of the Schistosoma species. Of those, 270,000 people (4.6%) suffer from pulmonary arterial hypertension, which is associated with the hepatosplenic form of the disease. This high prevalence makes schistosomiasis-associated pulmonary hypertension the leading cause of pulmonary hypertension worldwide. However, no specific treatment for the pulmonary vascular component of the disease has yet been devised. We report the case of a patient with schistosomiasis-associated pulmonary hypertension who was treated satisfactorily with a phosphodiesterase-5 inhibitor (sildenafil).
Keywords: Schistosomiasis mansoni; Hypertension, pulmonary; Drug therapy.
RESUMO
A esquistossomose mansoni é a terceira doença parasitária endêmica mais prevalente do mundo. Estima-se que mais de 200 milhões de pessoas estejam infectadas com uma das espécies do parasita Schistosoma. Dessas, 270.000 pessoas (4,6%) são portadoras de hipertensão arterial pulmonar, que é associada à forma hepatoesplênica da doença. Essa alta prevalência coloca a hipertensão pulmonar esquistossomótica como a causa mais frequente de hipertensão pulmonar no mundo. Entretanto, o tratamento dirigido especificamente ao acometimento vascular pulmonar não está ainda estabelecido. Relatamos o caso de uma paciente portadora dessa doença que foi tratada com um inibidor de fosfodiesterase-5 (sildenafil) com resultados satisfatórios.
Palavras-chave: Esquistossomose mansoni; Hipertensão pulmonar; Quimioterapia.
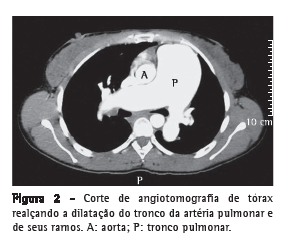
Introdução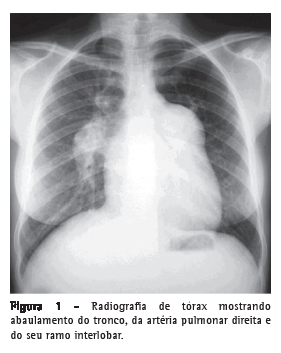



Sociedade Brasileira de Pneumologia e Tisiologia
Secretaria do Jornal Brasileiro de Pneumologia
SCS Quadra 01, Bloco K, Salas 203/204 Ed. Denasa
Brasília - DF CEP: 70.398-900
Fone/fax: 08000 61 6218, (61) 3245 1030 e (61) 3245 6218
E-mail: jbp@sbpt.org.br
Development by: ![]()
© All rights reserved 2025 - Jornal Brasileiro de Pneumologia
 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket




