ABSTRACT
Objective: To evaluate the association between nutritional status measurements and pulmonary function in children and adolescents with cystic fibrosis. Methods: We evaluated the nutritional status of 48 children and adolescents (aged 6-18 years) with cystic fibrosis based on body mass index (BMI) and body composition measurements-mid-arm muscle circumference (MAMC) and triceps skinfold thickness (TST)-at a referral center in the city of Rio de Janeiro, Brazil. Pulmonary function was assessed by means of spirometry, using FEV1 to classify the severity of airway obstruction. We used Student's t-tests for comparisons between proportions and linear regression analysis for associations between continuous variables. The level of significance was set at p < 0.05. Results: The evaluation of nutritional status based on BMI identified a smaller number of malnourished patients than did that based on MAMC (14 vs. 25 patients, respectively). Most of the patients presented mild pulmonary disease. Mean FEV1 was 82.5% of predicted. Pulmonary function was found to correlate significantly with BMI, MAMC and TST (p = 0.001, p = 0.001 and p = 0.03, respectively). All subjects with moderate or severe pulmonary involvement were considered malnourished based on BMI and body composition parameters. Of the 25 patients considered malnourished based on body composition (MAMC), 19 were considered well-nourished based on their BMI. Conclusions: In the present study, all nutritional status measurements correlated directly with the pulmonary function of children and adolescents with cystic fibrosis. However, body composition measurements allowed earlier detection of nutritional deficiencies.
Keywords:
Cystic fibrosis; Body composition; Spirometry; Nutrition assessment.
RESUMO
Objetivo: Avaliar a associação de medidas do estado nutricional com a função pulmonar de crianças e adolescentes com fibrose cística. Métodos: Foi avaliado o estado nutricional pelo índice de massa corpórea (IMC) e por medidas de composição corporal-circunferência muscular do braço (CMB) e medida da dobra cutânea triciptal (DCT)-de 48 crianças e adolescentes (6-18 anos) com fibrose cística em um centro de referência na cidade do Rio de Janeiro. A função pulmonar foi analisada por espirometria, e o parâmetro adotado para a classificação da obstrução das vias aéreas foi VEF1. Utilizou-se o teste t de Student para a comparação entre proporções e a regressão linear para associação entre variáveis contínuas. O nível de significância adotado foi p < 0,05. Resultados: A avaliação do estado nutricional através do IMC encontrou menos pacientes desnutridos do que a realizada através da CMB (14 vs. 25, respectivamente) A maioria dos pacientes apresentou doença pulmonar leve. A média do VEF1 foi de 82,5% do previsto. Houve associação do IMC, da CMB e da DCT com a função pulmonar (p = 0,001, p = 0,001 e p = 0,03, respectivamente). Todos os pacientes com comprometimento moderado e grave pulmonar eram desnutridos pela composição corporal (CMB). Entre os 25 pacientes considerados desnutridos através da composição corporal (CMB), 19 apresentavam peso adequado quando avaliados pelo IMC. Conclusões: Todas as medidas do estado nutricional apresentaram associação direta com a função pulmonar das crianças e adolescentes com fibrose cística no estudo. Entretanto, as medidas de composição corporal revelaram, de forma mais precoce, possíveis deficiências nutricionais.
Palavras-chave:
Fibrose cística; Composição corporal; Espirometria; Avaliação nutricional.
IntroduçãoA fibrose cística (FC) é uma doença genética autossômica recessiva caracterizada por uma grande variedade de manifestações clínicas. Dentre elas, as principais são DPOC supurativa progressiva, insuficiência pancreática, desnutrição secundária, aumento das concentrações de cloro e de sódio no suor e infertilidade masculina na idade adulta.(1)
A infecção bacteriana crônica do trato respiratório causa destruição e perda da função pulmonar, e a doença respiratória é uma das principais causas das limitações para a longevidade e do aumento da frequência das internações destes pacientes.(2)
A manutenção de um estado nutricional adequado é fundamental para a integridade do sistema respiratório na FC.(3,4) Uma das principais causas de depleção nutricional é o aumento do gasto energético devido à inflamação e à infecção pulmonar,(3) caracterizando a estreita relação entre o estado nutricional e a função pulmonar.
Os distúrbios minerais, eletrolíticos e energéticos, em nível muscular, são responsáveis pela disfunção e pela diminuição da contratilidade da musculatura respiratória acessória e do diafragma. A desnutrição pode comprometer o parênquima, a resposta imunológica pulmonar e o comando ventilatório.(5) Esse quadro culmina em diminuição do desempenho respiratório ao esforço, aumento da suscetibilidade às infecções pulmonares e falência respiratória.(6)
Um estudo realizado sobre turnover proteico em adultos sugeriu que pessoas com FC estão em um estado crônico de estresse catabólico ou de desnutrição ou ambos, ligado a recorrentes exacerbações pulmonares que afetam adversamente o balanço energético e o metabolismo proteico.(7)
Nos últimos anos, a importância da nutrição na evolução da FC é destacada, em diversos estudos, como essencial na determinação do prognóstico da doença.(4,5,7,8)
A utilização associada de critérios antropométricos, a fim de quantificar e qualificar o estado nutricional de forma mais precisa, torna-se essencial para que se possa diagnosticar e intervir adequadamente nas mudanças ou alterações do estado nutricional da forma mais precoce possível.
Dessa forma, o objetivo deste estudo foi avaliar a associação entre o estado nutricional, determinado através de índices antropométricos e de composição corporal, e a função pulmonar de crianças e adolescentes com FC acompanhados em um centro de referência do Rio de Janeiro.
MétodosTrata-se de um estudo descritivo transversal com 48 crianças e adolescentes com FC, segundo os critérios diagnósticos do consenso da Cystic Fibrosis Foundation.(9) Os critérios de inclusão para o estudo foram os seguintes: ter idade entre 6 e 18 anos e estar em acompanhamento no Ambulatório de Nutrição do Instituto Fernandes Figueira (IFF) da Fundação Oswaldo Cruz. Pacientes com diabetes ou com doença hepática foram excluídos do estudo. O projeto de pesquisa foi aprovado pelo Comitê de Ética e Pesquisa do IFF, e todos os responsáveis pelos pacientes assinaram o termo de consentimento livre e informado.
Os dados antropométricos e de função pulmonar foram coletados entre janeiro de 2006 e janeiro de 2008.
O peso (kg) foi aferido sem sapatos e com o mínimo de roupas em uma balança antropométrica do tipo plataforma (Filizola, São Paulo, Brasil) e a estatura (cm) em um estadiômetro acoplado à balança, com a posição da cabeça ajustada ao plano de Frankfurt. O índice de massa corpórea (IMC) foi calculado pelo peso dividido pela altura elevada ao quadrado (kg/m2).
A circunferência de braço (CB) foi medida no ponto médio do braço direito, relaxado, entre o ponto acromial da escápula e o olecrano da ulna. A dobra cutânea de tríceps (DCT) foi aferida com adipômetro Lange® (Cambridge Scientific Industries, Cambridge, MD, EUA) na face posterior do ponto médio do braço direito (mm), por três vezes, sendo considerado o valor médio entre as três medidas.
A circunferência muscular do braço (CMB) foi calculada pela subtração do valor da CB (cm) pelo produto entre o valor da DCT (convertida para cm) e o valor de ω, conforme a seguinte equação(10):
CMB(cm) = CB(cm) − (DCT(cm) × ω)
O referencial populacional para a avaliação nutricional foi o gráfico do National Center for Health Statistics, 2000.(11) A análise dos dados da avaliação nutricional foi realizada em percentis, de acordo com o Consenso em Nutrição de Pacientes Pediátricos com Fibrose Cística.(7)
Foram considerados desnutridos os pacientes que apresentassem IMC para idade (IMC/I) menor que o percentil 10; sob risco nutricional, aqueles com IMC/I entre o percentil 10 e o 25; e nutridos aqueles com IMC/I maior que o percentil 25. Para a classificação de baixa estatura, consideraram-se os valores de estatura para idade menores que o percentil 5.(7)
A CMB e a DCT foram analisadas de acordo com Frisancho,(10) sendo considerados baixos os valores menores ou iguais ao percentil 5. A análise do estágio puberal foi realizada segundo os critérios de Tanner.(12)
A avaliação da função pulmonar foi realizada pelo Setor de Provas de Função Respiratória do IFF. Para a validade dos resultados, estabeleceu-se a faixa etária mínima de 6 anos de idade, pois a espirometria depende da cooperação do paciente. O VEF1 foi utilizado para a classificação do comprometimento pulmonar quanto à obstrução. A técnica de realização do exame e os valores de referência seguiram as recomendações da American Thoracic Society, que classifica os valores percentuais do previsto da seguinte forma: obstrução leve, ≥ 70%; obstrução moderada, 60-69%; obstrução moderadamente severa, 50-59%; obstrução severa, 35-49%; e obstrução muito severa, < 35%.(13) Os pacientes com sobrepeso foram excluídos do estudo.
Os dados foram analisados no programa estatístico Statistical Package for the Social Sciences, versão 8.0 para Windows (SPSS Inc., Chicago, IL, EUA). Utilizou-se o teste t de Student para a comparação de proporções e a regressão linear para a associação entre variáveis contínuas. O nível de significância adotado foi de p < 0,05.
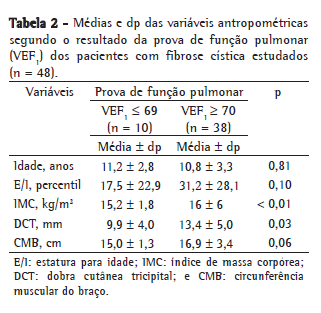
ResultadosEntre os 48 pacientes estudados, 29 eram do sexo feminino e 19 do sexo masculino. A média de idade foi de 10,8 ± 3,3 anos (Tabela 1). A maioria dos indivíduos (79%) possuía comprometimento leve da função pulmonar (Tabela 2).
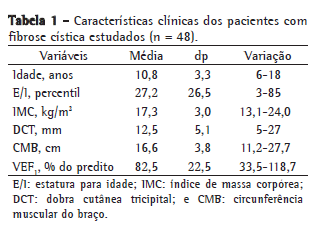
Nenhum paciente apresentou diminuição do depósito de gordura pela aferição da DCT. A prevalência de baixa estatura no grupo foi de 19,6%, e nenhum paciente apresentou atraso da puberdade.
A avaliação nutricional, utilizando-se como critério a CMB, demonstrou um maior percentual de desnutrição (52%) quando comparada à avaliação utilizando-se o IMC/I, que demonstrou 31,2% de participantes desnutridos e 14,5% com risco nutricional (Tabela 3). Dos pacientes considerados desnutridos segundo a CMB, 6 foram classificados como sob risco nutricional e 39% como nutridos quando avaliados pelo IMC/I.
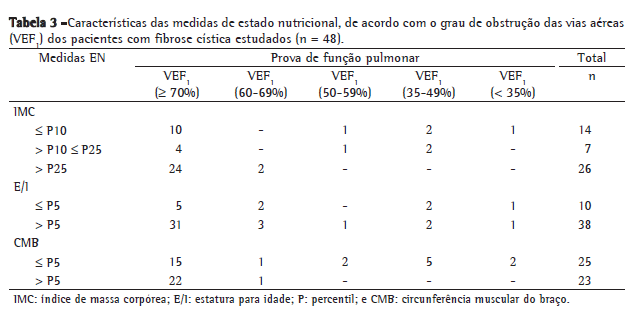
Todos os pacientes com obstrução moderada ou grave da função pulmonar foram considerados desnutridos utilizando-se como critério a CMB (Tabela 3).
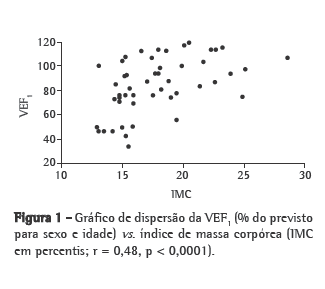
O IMC e a DCT apresentaram uma associação estatisticamente significativa (p = 0,001 e p = 0,03, respectivamente) com o grau de acometimento pulmonar, enquanto a CMB apresentou apenas uma tendência (p = 0,06) para esta mesma associação (Tabela 2). A regressão linear demonstrou uma relação moderada entre IMC e VEF1 (r = 0,46; p = 0,00001) e entre VEF1 e CMB (r = 0,46; p = 0,0001; Figuras 1 e 2).
 Discussão
DiscussãoO estudo encontrou maior prevalência de desnutrição utilizando-se a CMB como critério do que através do IMC/I, apesar deste apresentar um ponto de corte para a desnutrição maior em pacientes com FC (percentil 10) do que para a população geral (percentil 5). Entre os pacientes desnutridos, utilizando-se a CMB, 39% destes apresentaram-se eutróficos quando avaliados utilizando-se o IMC/I. Estes dados evidenciam a relevância da análise da composição corporal para uma melhor avaliação do estado nutricional.
A relação moderada encontrada entre o estado nutricional e a VEF1 (Figuras 1 e 2) nos pacientes deste estudo provavelmente ocorreu porque a queda da função pulmonar não é explicada unicamente pelo aumento do comprometimento do estado nutricional. As faltas de aderência ao tratamento e à fisioterapia respiratória regular são exemplos de outros fatores que interferem na manutenção da função pulmonar.
O comprometimento leve da função pulmonar apresentado pela maioria dos pacientes do estudo pode ter influenciado a tendência (p < 0,06) de associação entre a função pulmonar e a avaliação do estado nutricional utilizando-se a CMB, porque, nos casos de pouco comprometimento pulmonar, a depleção nutricional ocorre por outras causas, tais como o tipo de mutação genética, a presença de insuficiência pancreática, de perda de sais e ácidos biliares, de refluxo gastresofágico, assim como a ocorrência de ressecção intestinal e as condições emocionais dos pacientes. Desta forma, a fisiopatologia da desnutrição encontrada neste estudo se assemelha à desnutrição primária, que poupa a massa muscular em detrimento da massa gorda.
A adiposidade apresentou uma associação estatisticamente significativa com a função pulmonar (p = 0,03), embora a medida de DCT de todos os pacientes estivesse dentro da faixa de normalidade (Tabela 2). Talvez o ponto de corte para DCT (percentil 5) deva ser maior para os pacientes com FC. Alguns autores também encontraram uma associação positiva entre a adiposidade e a função pulmonar após estudar a composição corporal de 50 pacientes pediátricos com FC por absorciometria de dupla energia.(6)
Todos os pacientes com obstrução moderada ou grave da função pulmonar possuíam desnutrição pelo critério da CMB (Tabela 3). Este resultado já era esperado, pois a literatura é unânime em afirmar que a inflamação e a infecção pulmonar aumentam o gasto energético e se tornam uma das principais causas de depleção nutricional, principalmente de massa muscular, mesmo nos pacientes com ingestão adequada ou acima do recomendado.(2,5,14,15)
A baixa estatura encontrada neste estudo poderia ser atribuída à influência genética, mas a ausência de registro de dados para o cálculo do alvo genético não nos permite tal afirmação. Estudos com crianças brasileiras e norte-americanas encontraram resultados semelhantes e mostraram que a baixa estatura em pacientes com FC independe do nível do desenvolvimento do país e da carga genética, mas se relaciona a fatores inerentes à fisiopatologia da doença.(16,17)
O declínio da função pulmonar com o passar da idade é universal na FC.(18) Entretanto, no presente estudo, a média de idade dos pacientes entre os dois grupos separados pelo grau de comprometimento pulmonar foi semelhante.
Neste trabalho, 10 pacientes (19,6%) apresentaram baixa estatura; destes, 60% tinham comprometimento moderado ou grave da função pulmonar. Este resultado está de acordo com os dados de um relatório da Cystic Fibrosis Foundation,(19) que demonstraram índices de estatura para idade abaixo do percentil 5 em 20% das crianças. Há muito tempo se reconhece que a deficiência nutricional pode afetar significativamente a função pulmonar e que a doença pulmonar crônica pode levar ao déficit de crescimento e ao atraso do desenvolvimento, criando um círculo vicioso entre desnutrição e distúrbios respiratórios.(14,20)
Vários estudos demonstram que o IMC é um método mais sensível do que o índice de peso para estatura.(4,17,21,22) Neste estudo, ele apresentou uma associação estatisticamente significativa com a função pulmonar. Porém, esta medida não deve ser usada isoladamente para avaliar o estado nutricional desses pacientes, porque não determina a composição corporal.
Na FC, o desequilíbrio entre a síntese e a degradação proteica para suprir as necessidades energéticas do organismo pode causar uma depleção desproporcional da massa magra, com atrofia da musculatura, inclusive a respiratória.(18) Desta forma, a alteração da composição corporal pode levar à desnutrição e à diminuição da função pulmonar.(6,7,23)
Uma vez que o peso corpóreo é composto principalmente de massa gorda e massa livre de gordura, a observação em separado desses dois compartimentos corpóreos pode fornecer informações mais precisas sobre o estado nutricional, que está diretamente relacionado com a doença pulmonar-a principal causa de morbidade e mortalidade na FC.
Os resultados deste estudo apontam a alteração da composição corporal antes da diminuição do peso e da estatura de crianças e adolescentes com FC. Como a avaliação do estado nutricional, incluindo a medida das reservas adiposa e muscular através da DCT e da CB, é um método de fácil execução e baixo custo, os profissionais de saúde devem ser estimulados a utilizá-la durante as visitas de rotina, identificando mais precocemente o paciente em risco de desnutrição, para a indicação imediata da intervenção nutricional e, possivelmente, retardando a progressão da doença pulmonar.
As medidas de avaliação do estado nutricional apresentaram associação com a função pulmonar em crianças e adolescentes com FC neste estudo. Entretanto, a composição corporal nos revelou, de forma mais imediata, possíveis deficiências nutricionais. Assim, a associação de medidas antropométricas (IMC com a composição corporal) aumenta a acurácia no diagnóstico nutricional e detecta alterações precoces do estado nutricional, antecipando as intervenções nutricionais e retardando o declínio da função pulmonar destes pacientes.
Referências 1. Rozov T, Cunha MT, Nascimento O, Quittner AL, Jardim JR. Linguistic validation of cystic fibrosis quality of life questionnaires. J Pediatr (Rio J). 2006;82(2):151-6.
2. Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med. 1996;154(5):1229-56.
3. Peterson ML, Jacobs DR Jr, Milla CE. Longitudinal changes in growth parameters are correlated with changes in pulmonary function in children with cystic fibrosis. Pediatrics. 2003;112(3 Pt 1):588-92.
4. Stapleton D, Kerr D, Gurrin L, Sherriff J, Sly P. Height and weight fail to detect early signs of malnutrition in children with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2001;33(3):319-25.
5. Steinkamp G, Wiedemann B. Relationship between nutritional status and lung function in cystic fibrosis: cross sectional and longitudinal analyses from the German CF quality assurance (CFQA) project. Thorax. 2002;57(7):596-601.
6. Pedreira CC, Robert RG, Dalton V, Oliver MR, Carlin JB, Robinson P, et al. Association of body composition and lung function in children with cystic fibrosis. Pediatr Pulmonol. 2005;39(3):276-80.
7. Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr. 2002;35(3):246-59.
8. Thomson MA, Quirk P, Swanson CE, Thomas BJ, Holt TL, Francis PJ, et al. Nutritional growth retardation is associated with defective lung growth in cystic fibrosis: a preventable determinant of progressive pulmonary dysfunction. Nutrition. 1995;11(4):350-4.
9. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132(4):589-95.
10. Frisancho AR. New norms of upper limb fat and muscle areas for assessment of nutritional status. Am J Clin Nutr. 1981;34(11):2540-5.
11. Centers for Disease Control and Prevention [homepage on the Internet]. Atlanta: National Center for Health Statistics; c2000-2004 [updated 2001 Apr 20; cited 2004 Feb 10]. 2000 CDC Growth Charts: United States; [about 2 screens]. Available from: http://www.cdc.gov/growthcharts
12. Tanner JM. Growth at adolescence. Oxford: Blackwell Scientific Publications; 1962.
13. Pellegrino R, Viegi G, Brusasco V, Crapo RO, Burgos F, Casaburi R, et al. Interpretative strategies for lung function tests. Eur Respir J. 2005;26(5):948-68.
14. Pencharz PB, Durie PR. Pathogenesis of malnutrition in cystic fibrosis, and its treatment. Clin Nutr. 2000;19(6):387-94.
15. Zemel BS, Jawad AF, FitzSimmons S, Stallings VA. Longitudinal relationship among growth, nutritional status, and pulmonary function in children with cystic fibrosis: analysis of the Cystic Fibrosis Foundation National CF Patient Registry. J Pediatr. 2000;137(3):374-80.
16. Adde FV, Rodrigues JC, Cardoso AL. Nutritional follow-up of cystic fibrosis patients: the role of nutrition education. J Pediatr (Rio J). 2004;80(6):475-82.
17. Lai HC, Kosorok MR, Sondel SA, Chen ST, FitzSimmons SC, Green CG, et al. Growth status in children with cystic fibrosis based on the National Cystic Fibrosis Patient Registry data: evaluation of various criteria used to identify malnutrition. J Pediatr. 1998;132(3 Pt 1):478-85.
18. Dornelas EC, Fernandes MI, Galvão LC, Silva GA. Estudo do quadro pulmonar de pacientes com fibrose cística. J Pediatr (Rio J). 2000;76(4):295-9.
19. Cystic Fibrosis Foundation. Patient Registry 2002 Annual Data Report. Bethesda (MD): Cystic Fibrosis Foundation; 2003.
20. Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr. 2003;142(6):624-30.
21. Wiedemann B, Paul KD, Stern M, Wagner TO, Hirche TO; German CFQA Group. Evaluation of body mass index percentiles for assessment of malnutrition in children with cystic fibrosis. Eur J Clin Nutr. 2007;61(6):759-68.
22. Zhang Z, Lai HJ. Comparison of the use of body mass index percentiles and percentage of ideal body weight to screen for malnutrition in children with cystic fibrosis. Am J Clin Nutr. 2004;80(4):982-91.
23. Schöni MH, Casaulta-Aebischer C. Nutrition and lung function in cystic fibrosis patients: review. Clin Nutr. 2000;19(2):79-85.
Sobre os autores Célia Regina Moutinho de Miranda Chaves
Chefe do Departamento de Alimentação e Nutrição. Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz - Rio de Janeiro (RJ) Brasil.
José Augusto Alves de Britto
Médico Pediatra. Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz - Rio de Janeiro (RJ) Brasil.
Cristiano Queiroz de Oliveira
Preparador Físico. Departamento de Alimentação e Nutrição, Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz - Rio de Janeiro (RJ) Brasil.
Miriam Martins Gomes
Nutricionista. Departamento de Alimentação e Nutrição, Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz - Rio de Janeiro (RJ) Brasil.
Ana Lúcia Pereira da Cunha
Nutricionista. Departamento de Alimentação e Nutrição, Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz - Rio de Janeiro (RJ) Brasil.
Trabalho realizado no Instituto Fernandes Figueira/Fundação Oswaldo Cruz - IFF/Fiocruz - Rio de Janeiro (RJ) Brasil.
Endereço para correspondência: Célia Regina Murtinho de Miranda Chaves. Av. Rui Barbosa, 716, Flamengo, CEP 22250-020, Rio de Janeiro, RJ, Brasil.
Tel 55 21 2554-1718. E-mail: crchaves@iff.fiocruz.br
Apoio financeiro: Nenhum.
Recebido para publicação em 25/8/2008. Aprovado, após revisão, em 19/11/2008.












 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket