ABSTRACT
This article aims to review the physiopathology, diagnosis and treatment of cystic fibrosis-related dyslipidemia (CFD). Bibliographic searches of the Medline and Latin American and Caribbean Health Sciences Literature databases were made (year range, 1987-2007), and the most representative papers on the theme were selected. The characteristic symptoms of CFD are hypertriglyceridemia-with or without hypocholesterolemia-and essential fatty acid deficiency. The principal CFD risk factors are pancreatic insufficiency, high-carbohydrate diet, liver diseases, inflammatory state and corticosteroid therapy. There are no specific recommendations regarding screening, which is typically performed based on the diagnosis, and at regular intervals, and more frequently in individuals belonging to high-risk groups. Treatment includes a balanced diet, micronutrient supplementation, and regular physical exercise according to individual tolerance. In the great majority of the cases, CFD-related hypertriglyceridemia does not reach values for which the use of hypolipidemic drugs is indicated. We conclude that there are few articles in the literature regarding the frequency, etiology and management of CFD. Preventive and therapeutic recommendations for hypertriglyceridemia are extrapolated from studies in individuals without cystic fibrosis. Further research is necessary to investigate the association of essential fatty acid deficiency and the physiopathology of cystic fibrosis. Since hypertriglyceridemia is an important risk factor for coronary artery disease, prospective studies will contribute for a better understanding of the natural history of this condition and define how to prevent and treat it.
Keywords:
Cystic fibrosis; Dyslipidemias; Hypertriglyceridemia; Fatty Acids, nonesterified.
RESUMO
Esse artigo tem por objetivo rever o conhecimento atual sobre a fisiopatologia, o diagnóstico e a abordagem da dislipidemia relacionada à
fibrose cística (DFC). A pesquisa bibliográfica utilizou os bancos de dados Medline e Literatura Latino-Americana e do Caribe em Ciências
da Saúde (1987-2007), selecionando os artigos mais relevantes sobre o tema. A DFC é caracterizada por hipertrigliceridemia e/ou hipocolesterolemia
e deficiência de ácidos graxos essenciais. Seus principais fatores de risco são: insuficiência pancreática, dieta rica em carboidratos,
hepatopatias, estado inflamatório e corticoterapia. Não existem recomendações específicas sobre a triagem, que habitualmente é realizada a
partir do diagnóstico e, em intervalos regulares, com maior freqüência, nos indivíduos pertencentes aos grupos de risco. O tratamento inclui:
dieta balanceada, reposição de micronutrientes, vitaminas e fibras, além de exercício físico regular de acordo com a tolerância individual. Na
grande maioria dos casos, a hipertrigliceridemia da DFC não atinge valores que indiquem o uso de hipolipemiantes. Conclui-se que existem
poucos trabalhos na literatura sobre a freqüência, etiologia e manejo da DFC. A recomendações preventivas e terapêuticas para a hipertrigliceridemia
são extrapoladas de diretrizes para indivíduos sem fibrose cística. Mais pesquisas são necessárias para investigar a associação
da deficiência de ácidos graxos essenciais com a fisiopatologia da fibrose cística. Como a hipertrigliceridemia é um importante fator de risco
para doença arterial coronariana, estudos prospectivos irão contribuir para o melhor entendimento da história natural dessa complicação
bem como definir maneiras de preveni-la e tratá-la.
Palavras-chave:
Fibrose cística; Dislipidemias; Hipertrigliceridemia; Ácidos graxos não-esterificados.
IntroduçãoA fibrose cística (FC) é um distúrbio hereditário multissistêmico caracterizado primariamente por insuficiência pancreática exócrina, obstrução e infecção das vias aéreas.(1) As manifestações clínicas podem ser explicadas, na sua maioria, pela inflamação crônica e pela disfun Como resultado do aumento da expectativa de vida devido aos avanços no tratamento, algumas complicações antes não observadas passaram a ser diagnosticadas, como, por exemplo: diabetes melito, doença hepática, osteoporose e, mais recentemente, dislipidemia.(4)
Alguns indivíduos com FC apresentam um padrão dislipidêmico caracterizado por hipertrigliceridemia e/ou hipocolesterolemia.(5) A alteração mais freqüente é a hipertrigliceridemia, apesar desses pacientes terem aumento do gasto energético, má absorção intestinal de gordura e redução da ingestão alimentar.(6) Embora a hipertrigliceridemia seja um dos fatores de risco para doença cardiovascular aterosclerótica, pouca atenção tem sido dada ao impacto dessa complicação na morbidade dos pacientes com FC. Isto se torna mais importante quando houver associação com o diabetes melito relacionado à FC, uma vez que a hiperglicemia contribui para a aterogênese através da glicação e peroxidação da low-density lipoprotein (LDL; lipoproteína de baixa densidade).(4,7)
Devido à escassez de trabalhos sobre o tema, o presente artigo tem por objetivo realizar uma revisão crítica sobre o conhecimento atual da fisiopatologia, diagnóstico e abordagem da dislipidemia em pacientes com fibrose cística. No presente estudo, esta complicação foi denominada dislipidemia relacionada à FC (DFC). A pesquisa bibliográfica utilizou os bancos de dados Medline e Literatura Latino-Americana e do Caribe em Ciências da Saúde, selecionando os artigos mais relevantes sobre o tema publicados no período de 1987 a 2007.
Metabolismo lipídico normalColesterol e triglicérides (TG; ou triacilglicerol) são os principais lipídios plasmáticos. O colesterol é fundamental para a síntese de hormônios esteróides, síntese de ácidos biliares e formação das membranas celulares. Os TG são a principal reserva energética do organismo. Os ácidos graxos essenciais (por ex., linoléico e linolênico), participam do desenvolvimento e funcionamento do sistema nervoso central, sistema imune e função vascular. Entretanto, por não serem produzidos de forma adequada pelas células dos mamíferos, a manutenção de seus níveis séricos é dependente de sua adequada ingestão e absorção.
Por serem praticamente insolúveis em água, o colesterol e TG são transportados no plasma associados a proteínas, formando complexos solúveis denominados lipoproteínas.(8) As lipoproteínas têm forma esférica e são formadas pelo núcleo e pela camada externa. O núcleo contém principalmente ésteres de colesterol e TG.(8) A camada externa é composta de fosfolípides e colesterol livre.(9) Na sua superfície, encontram-se as apoproteínas ou apolipoproteínas que se ligam a receptores específicos das células responsáveis pelo metabolismo das lipoproteínas.(8,10)
As lipoproteínas são classificadas conforme sua mobilidade eletroforética e densidade (quanto maior a quantidade de apoproteínas, maior a densidade; e quanto maior a quantidade de TG, menor a densidade).(9) As principais lipoproteínas são: quilomícrons (QM), very low-density lipoprotein (VLDL; lipoproteína de densidade muito baixa), lipoproteínas de densidade intermediária, low-density lipoprotein (LDL; lipoproteínas de baixa densidade) e high-density lipoprotein (HDL; lipoproteínas de alta densidade). A Tabela 1 mostra as propriedades das principais lipoproteínas.
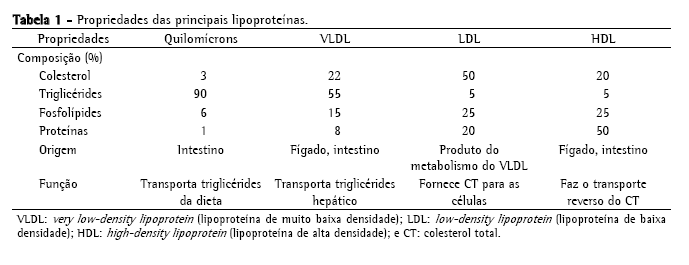
No intestino, o colesterol e TG da dieta são emulsificados por ácidos biliares e hidrolisados por lipases pancreáticas. Os TG são quebrados em ácidos graxos e monoglicérides, e os ésteres de colesterol em ácidos graxos e colesterol não esterificado. Na célula intestinal, os monoglicérides são re-esterificados em TG e armazenados nos QM. A ApoC-II, a ApoB-48 e a Apo-E são as principais apoliproteínas dos QM. Os QM são secretados na linfa através do ducto torácico e levados para a circulação sistêmica através da veia cava. No endotélio capilar, os QM se ligam à lipoproteína lipase através da ApoC-II, liberando os TG sob a forma de monoglicérides e ácidos graxos livres.(8) O músculo utiliza os ácidos graxos livres e monoglicérides para produzir energia. O tecido adiposo os utiliza para ressintetizar TG e armazená-los nas células adiposas.(9) O QM resultante desse metabolismo, QM remanescente, de menor tamanho e mais rico em colesterol, é captado pelo fígado. Normalmente, os QM só são encontrados no plasma no período pós-prandial, pois eles são os transportadores dos TG da dieta.(8) No fígado, ocorre a síntese de TG e colesterol, que são incorporados às VLDL, e depois excretadas.(10) A VLDL transporta TG do fígado para os tecidos periféricos. Na membrana celular, elas se ligam através da ApoC-II à lipoproteína lipase a fim de liberar os TG, formando as lipoproteínas de densidade intermediária.(8) As lipoproteínas de densidade intermediária são removidas pelo fígado via ApoE onde são transformadas em LDL. A LDL é a principal transportadora de colesterol para as células.(8) Nos tecidos extra-hepáticos, incluindo o espaço subendotelial onde o efeito protetor dos antioxidantes circulantes (por ex., vitamina E) é perdido, a LDL é oxidada, iniciando a formação das placas de ateroma. A HDL é produzida no fígado e intestino e atua no transporte reverso do colesterol, o processo em que a HDL capta o excesso de colesterol e TG liberados às células e os transporta para o fígado.(11)
Fisiopatologia da dislipidemia relacionada à fibrose císticaA fisiopatologia da DFC ainda não está totalmente elucidada. Os mecanismos mais estudados são: dieta rica em carboidratos e pobre em gorduras, dieta rica em gorduras, hepatopatia, elevação das citocinas pró-inflamatórias e uso de corticóides, provavelmente atuando de forma combinada.(5)
Estudos prévios não demonstram associação da dislipidemia com intolerância a glicose, diabetes melito, tireoidopatia, genótipos específicos da FC, função pulmonar, concentração da proteína C reativa, idade, sexo, peso, índice de massa corporal e pressão arterial.(5,12)
Dieta rica em carboidratos e pobre em gorduraPacientes com insuficiência pancreática exógena e reposição enzimática inadequada não toleram dietas com teor normal de gordura devido à má absorção intestinal, e, como mecanismo de defesa, consomem um excesso de carboidratos.(5,13) O consumo dessas dietas com baixo teor de gorduras e ricas em carboidratos pode ser uma das causas da hipertrigliceridemia relacionada à FC. Nesses casos, o consumo excessivo e crônico de carboidratos faz com que o fígado ultrapasse sua capacidade de síntese e armazenamento de glicogênio, desviando esta via metabólica para a produção de TG.(14) Outras explicações seriam a diminuição da atividade da lipoproteína lípase e aumento da absorção intestinal de glicose.(15)
Dieta rica em gorduraA fim de estudar a repercussão de uma dieta rica em gordura sobre os lipídios séricos, um estudo comparou o perfil lipídico de adultos fibrocísticos, com e sem insuficiência pancreática exógena, com controles saudáveis.(16)
Observou-se que pacientes com FC e insuficiência pancreática, apesar de consumirem grandes quantidades de gordura e/ou colesterol, não apresentaram piora do perfil lipídico, em contraste com aqueles sem insuficiência pancreática exógena, nos quais se observou o mesmo risco aterogênico da população geral. Ou seja, fibrocísticos com função pancreática exógena normal apresentam risco elevado de aterogênese, semelhante à população geral, ao consumirem dietas ricas em gordura. Entretanto, esta hipótese tem sido controversa desde que em um estudo não se observou diferença significativa no perfil lipídico de pacientes com FC quando comparados com o grupo controle, apesar do uso da dieta rica em gorduras,(17) e em outro estudo relatou-se que, mesmo com suplementação enzimática, a absorção de gordura da dieta variou de 79-93%.(18)
Disfunção hepáticaA doença hepática é considerada uma séria co-morbidade da FC, alterando seu prognóstico e a qualidade de vida.(19) A prevalência depende dos critérios utilizados: desde 1,4-7% por avaliação clínica, até mais de 20% se forem utilizados métodos bioquímicos e ultrassonográficos.(19) A hepatopatia é resultante da esteatose hepática, a qual é usualmente atribuída ao acúmulo de TG e LDL no parênquima hepático devido à redução na síntese de apolipoproteínas e, em alguns casos, devido à deficiência de carnitina.(20) Dependendo da sua gravidade, a hepatopatia pode contribuir para a
hipertrigliceridemia.(21)
Aumento dos níveis séricos do fator de necrose tumoral alfa e da proteína C reativaCitocinas pró-inflamatórias, como o fator de necrose tumoral alfa, são mediadores de hiperlipidemia em situações de infecção ou estresse ao inibirem a atividade da lipase lipoprotéica (diminuindo o clearance de TG) e estimularem a lipogênese hepática.(22) Portanto, a hipertrigliceridemia da FC pode ser relacionada ao estado inflamatório crônico exacerbado pelos freqüentes surtos de infecção. A proteína C reativa, outro mediador inflamatório, não se associou à dislipidemia.(5) Estudos recentes têm chamado atenção para a possibilidade de tratamentos com drogas antiinflamatórias sistêmicas serem benéficos para pacientes com doenças inflamatórias crônicas, como a FC, melhorando o estado nutricional e reduzindo a resistência insulínica, um dos fatores contribuintes para a dislipidemia.(2)
Alteração na absorção intestinal de ácidos graxos livresUm estudo observou a deficiência de ácidos graxos essenciais inclusive em fibrocísticos bem nutridos com consumo normal de gorduras e na ausência de manifestações clínicas de má absorção intestinal.(23) Os autores acreditam que tal problema ocorra devido à deficiência de micronutrientes (por ex., selênio e tocoferol), infecção crônica (que aumenta a produção de eicosanóides), defeito na incorporação de ácidos graxos na membrana celular ou a alterações no metabolismo lipoprotéico.
Em outro estudo observou-se que a alteração na absorção de gorduras nos pacientes fibrocísticos ocorre devido à solubilização intraluminal incompleta ou reduzida absorção dos ácidos graxos de cadeia longa pela mucosa intestinal, não havendo habitualmente insuficiente reposição enzimática.(18)
Embora a deficiência de ácidos graxos em diferentes células e tecidos tenha sido inicialmente atribuída apenas à má absorção intestinal de gordura,(24) atualmente se sabe que tal distúrbio é primário em alguns pacientes como resultado de alterações na CFTR.(25)
Uso de corticóidesCorticóides são uma das causas mais importantes de dislipidemia secundária, principalmente de hipertrigliceridemia.(26) Como os pacientes com FC, principalmente aqueles com complicações pulmonares, fazem uso mais freqüente e/ou crônico desses medicamentos, este pode ser, teoricamente, mais um fator contribuinte para a dislipidemia. Entretanto, não foram encontrados estudos na literatura mostrando essa associação em pacientes com FC.(5)
Alterações lipoprotéicas na fibrose císticaAlterações quantitativasAs principais alterações quantitativas do metabolismo lipídico na FC são a hipertrigliceridemia e hipocolesterolemia. Em pacientes norte-americanos com FC, na faixa etária dos 5 aos 19 anos, a média do colesterol sérico foi 133 ± 30 mg/dL e a de TG foi 126 ± 70 mg/dL em comparação com, respectivamente, 149 mg/dL e 55 mg/dL para a população da mesma idade e sexo sem FC. Na faixa etária dos 20 aos 44 anos, os valores médios para colesterol total (CT) e TG foram 155 ± 39 mg/dL e 162 ± 118 mg/dL, quando comparados, respectivamente, com 199 mg/dL e 87 mg/dL para o grupo controle.(5) Isto reforça os dados que fibrocísticos, apresentam, em média, valores de colesterol inferiores, e valores de TG superiores em relação a sua média populacional. Nesse mesmo estudo, hipertrigliceridemia definida por valores acima de 200 mg/dL foi encontrada apenas em 16% dos pacientes.
Alterações qualitativasAproximadamente 85% dos pacientes com FC apresentam deficiência de ácidos graxos essenciais.(27) O perfil típico desta deficiência é caracterizado por diminuição da concentração dos ácidos linoléico (18:2n-6) e docosaexaenóico (22:6n-3); e aumento do ácido eicosatrienóico (20:3n-9), ácido oléico (18:1n-9) e ácido palmitoléico (16:1n-7).(27-30) Como estas alterações também foram detectadas em pacientes com FC bem nutridos e sem insuficiência pancreática exógena e em seus pais, isto sugere que possa existir um defeito no metabolismo dos ácidos graxos relacionado à etiologia da FC, como, por exemplo, aumento de sua oxidação como fonte de energia, utilização como precursores da resposta inflamatória, metabolismo deficiente e alterações da CFTR.(25,27) Ainda não se sabe como esse perfil de ácidos graxos se relaciona a hipocolesterolemia e hipertrigliceridemia dos pacientes com FC.
Deficiência de ácidos graxos essenciais e inflamação na fibrose císticaA morbidade e a mortalidade dos pacientes com FC é em grande parte resultado da doença pulmonar, a qual, por sua vez, é caracterizada por aumento da resposta inflamatória mediada por neutrófilos, linfócitos e citocinas pró-inflamatórias.(31) Pacientes com FC apresentam aumento da concentração de ácido aracdônico e diminuição da concentração do ácido docosaexaenóico.(24-25) O ácido aracdônico é um substrato para síntese de prostaglandina E2, tromboxano A2 e leucotrieno B4, contribuindo assim para o processo inflamatório típico da doença.(24) O ácido docosaexaenóico é convertido em potentes mediadores anti-inflamatórios (docosatrienos), fazendo com que a redução dos seus níveis séricos possa contribuir para um aumento da resposta inflamatória.(32) Além disso, as alterações dos ácidos graxos essenciais na FC podem causar diminuição da expressão e/ou atividade do peroxisome proliferator-activated receptor alpha (PPARα; receptor ativado por proliferadores de perosissoma alfa) em linfócitos. Como o PPAR-α exibe atividade antiinflamatória, sua diminuição, em pacientes com FC, poderia ser outro mecanismo responsável pela resposta inflamatória exacerbada.(31) Tal observação é importante pois pode conduzir a pesquisas que usem fibratos e glitazonas, ativadores sintéticos do PPARα, no tratamento da FC.
Assim, caso seja possível correlacionar os níveis séricos dos ácidos graxos essenciais com suas concentrações teciduais, talvez seja razoável tentar normalizá-los com o objetivo de atenuar a resposta inflamatória crônica. Corroborando esses dados, um estudo(33) mostrou que os níveis de ácidos graxos aumentavam em pacientes com FC após tratamento com antibióticos, e em outro estudo(34) relatou-se melhora da resposta inflamatória em camundongos infectados cronicamente por Pseudomonas aeruginosa após suplementação com ácidos graxos ômega-3.
Dislipidemia e risco cardiovascular na fibrose císticaAinda não está claro o papel da hipertrigliceridemia como fator de risco independente para doença cardiovascular.(26) Em indivíduos sem FC, esse risco estaria aumentado em situações nas quais a hipertrigliceridemia se associa à presença de frações de VLDL menores e ricas em colesterol (por ex., diabetes melito, obesidade e síndrome metabólica); e seria inexistente nas hipertrigliceridemias acompanhadas de frações de VLDL grandes e ricas em TG (por ex., dietas ricas em carboidratos e ingestão excessiva de álcool).(8)
Como pacientes com FC e hipertrigliceridemia não apresentam fatores de risco aterogênicos (por ex., obesidade, síndrome metabólica), exceto pela presença de diabetes melito em alguns casos, e como a dieta rica em carboidratos não se associa a um maior risco de doença cardiovascular, fica ainda mais difícil avaliar a repercussão cardiovascular da DFC. Um estudo epidemiológico recente mostrou, na população em geral, a existência de uma associação moderada, mas bastante significante, entre níveis elevados de TG e risco aumentado de doença coronariana.(35) Tal fato chama atenção para a necessidade de avaliar esse risco em pacientes com FC e hipertrigliceridemia.
Abordagem da dislipidemia relacionada à fibrose císticaTriagem e diagnósticoNão existe uma recomendação especifica para triagem e diagnóstico laboratorial da DFC. Por isto, as orientações e valores de referência são extrapolados das diretrizes da National Cholesterol Education Program, de 1992(36) e da Sociedade Brasileira de Cardiologia, de 2005(37) para crianças (Tabela 2); e das recomendações do Adult Panel III of the National Cholesterol Education Program(38) (Tabela 3) para adultos. Os valores de referência sugeridos pela Sociedade Brasileira de Cardiologia para indivíduos menores de 19 anos diferem daqueles da National Cholesterol Education Program, uma vez que propõem valores inferiores de CT, LDL e TG.

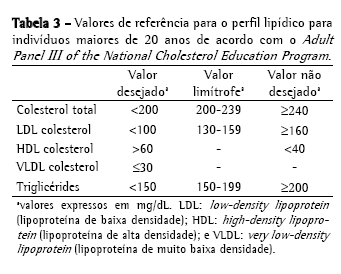
A triagem da dislipidemia é realizada através da determinação do perfil lipídico (CT, HDL, VLDL e TG) após 12 horas de jejum. O LDL é calculado pela fórmula de Friedewald (LDL = [CT − HDL] − [TG/5]) se os TG < 400 mg/dL. Idealmente, o perfil lipídico deverá ser determinado em indivíduos usando sua dieta habitual, na ausência de grandes variações recentes no peso e sem terem realizado atividade física vigorosa ou consumido álcool nas 24 h que antecedem o exame. Deve-se questionar sobre uso de medicamentos que alteram o perfil lipídico como corticóides, contraceptivos hormonais, anti-hipertensivos e anticonvulsivantes e sobre os fatores de risco tradicionais para dislipidemia como tabagismo, hipertensão e sedentarismo, entre outros.(26)
Resultados anormais devem ser confirmados por um segundo exame. Se persistirem alterados é fundamental excluir e tratar causas secundárias como hipotireoidismo, diabetes, nefropatia, sedentarismo, consumo de álcool e uso de medicamentos, principalmente corticóides e estrógenos. (8,26)
Apesar dos relatos freqüentes de deficiência de ácidos graxos essenciais, principalmente o linoléico e o docosaexaenóico, não existem recomendações quanto à dosagem rotineira dos ácidos graxos livres no sangue ou nas membranas dos eritrócitos de pacientes com FC.(30) Alguns dos fatores que dificultam o estabelecimento de um valor de normalidade são: infecções (diminuem os níveis de ácidos graxos livres), estado nutricional, severidade da mutação do CFTR e o fato dos níveis séricos de ácidos graxos não refletirem sua concentração tecidual.(25,27,33)
TratamentoNão-farmacológicoDevido à maior necessidade metabólica como resultado do elevado gasto energético basal, perda de gordura pela má absorção intestinal de gorduras e redução da ingestão alimentar, especialmente durante os episódios de infecção, pacientes com FC são aconselhados a fazer uso de dieta balanceada, hiperprotéica e hipercalórica (120-150% das recomendações diárias tradicionais), com suplementação de micronutrientes de acordo com suas deficiências específicas e reposição de vitaminas lipossolúveis, assim como fibras.(2,6)
Na ausência de insuficiência pancreática exógena, a recomendação de energia deve ser semelhante à população sem FC, por isso o uso de dieta hipercalórica com alto teor de lípides deve ser monitorado pelo risco de dislipidemia.(16) Se houver insuficiência pancreática exógena, a suplementação enzimática é necessária. Não se deve esperar pela normalização de gorduras nas fezes, uma vez que o uso de doses excessivas de enzimas não traz maiores benefícios e pode causar dano à estrutura do intestino levando a uma colonopatia fibrosante.(6,18) A dose máxima de enzimas pancreáticas não deve exceder 10.000 UI lipase/kg/dia.(39)
Em pacientes sem FC, as recomendações dietéticas para o tratamento da hipertrigliceridemia incluem restrição da ingestão de gordura saturada e trans e aumento do consumo de ácidos graxos ômega-3.(26) A restrição de gorduras para menos de 30% do consumo energético total não se aplica à FC, uma vez que a gordura representa 31-35% da sua ingestão calórica desde que, mesmo com reposição de enzimas pancreáticas, 5-20% da gordura da dieta não seja absorvida.(18) Em um estudo,(40) relatou-se maior ingestão de gordura saturada e monoinsaturada e menor ingestão de gordura polinsaturada em pacientes com FC quando comparados aos controles, chamando atenção para a necessidade de se balancear a ingestão de gordura nesses pacientes, a fim de minimizar a deficiência de ácidos graxos essenciais.
Exercícios físicos regulares vêm sendo considerados como parte fundamental do manejo do paciente portador de FC, melhorando a performance cardiorrespiratória, força muscular, aquisição de massa óssea e percepção corporal. Os exercícios devem ser individualizados, respeitando a capacidade e a tolerância de cada paciente.(41)
FarmacológicoAs recomendações para o tratamento da hipertrigliceridemia não são tão claras quanto para o tratamento da hipercolesterolemia. Hipolipemiantes podem ser recomendados para hipertrigliceridemias graves (>500 ou 1000 mg/dL) que não respondem ao tratamento não farmacológico devido ao elevado risco de pancreatite.(8,26)
Os fibratos (gemfibrozil, bezafibrato, fenofibrato) são as drogas de primeira escolha para o tratamento da hipertrigliceridemia.(8) Seu principal mecanismo de ação ocorre através da modulação da atividade do PPAR-α no fígado, com redução da secreção hepática de VLDL e TG e aumento da lipólise de TG plasmáticos.(26) Porém, seu uso na faixa etária pediátrica aguarda mais experiência.(37)
Em relação aos outros hipolipemiantes, as estatinas (reduzem a produção hepática de colesterol) e ezetimiba (reduz a absorção intestinal do colesterol) não são indicados, porque uma das características da DFC é a hipocolesterolemia. As resinas seqüestradoras de ácidos biliares (colestipol e colestiramina) não são indicadas por poderem aumentar os TG (maior síntese de VLDL) e por contribuírem para a má absorção intestinal.(37)
Alguns estudos tem sido realizados com o ácido ursodeoxicólico com o objetivo de melhorar o fluxo biliar e assim prevenir ou tratar a doença hepática relacionada à FC e, desse modo, reduzir o risco de dislipidemia.(42)
Entretanto, em um estudo,(43) não se observaram evidências significativas de melhora da função hepática com o uso de tal medicação.
O uso de suplementação com ácidos graxos ômega-3 e ômega-6 tem apresentado resultados discrepantes, sendo necessária a realização de mais estudos para a recomendação de seu uso no tratamento da FC.(27,30,34)
Considerações finais e perspectivas para o futuroExistem poucos estudos avaliando a prevalência e a fisiopatologia da DFC. Por isto, não existem consensos orientando como realizar a triagem para esse distúrbio, como preveni-lo, e, se presente, como tratá-lo. Devido ao aumento da expectativa de vida dos fibrocísticos e diante das alterações metabólicas apresentadas por esses indivíduos, é fundamental a realização de trabalhos prospectivos visando responder a maioria dessas questões, principalmente o papel da hipertrigliceridemia isolada como fator de risco para doenças cardiovasculares. No momento, a maioria das recomendações quanto ao manejo da hipertrigliceridemia provém de estudos em pacientes sem FC e com outros fatores de risco cardiovasculares. Mais pesquisas também são necessárias visando a esclarecer melhor o papel da deficiência de ácidos graxos essenciais na fisiopatologia da FC.
Referências 1. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998;132(4):589-95.
2. Elborn JS. How can we prevent multisystem complications of cystic fibrosis? Semin Respir Crit Care Med. 2007;28(3):303-11.
3. Alvarez AE, Ribeiro AF, Hessel G, Bertuzzo CS, Ribeiro JD. Cystic fibrosis at a Brazilian center of excellence: clinical and laboratory characteristics of 104 patients and their association with genotype and disease severity [Article in Portuguese]. J Pediatr (Rio J). 2004;80(5):371-9.
4. Alves CA, Aguiar RA, Alves AC, Santana MA. Diabetes melito: uma importante co-morbidade da fibrose cística. J Bras Pneumol. 2007; 33(2):213-21.
5. Figueroa V, Milla C, Parks EJ, Schwarzenberg SJ, Moran A. Abnormal lipid concentrations in cystic fibrosis. Am J Clin Nutr. 2002;75(6):1005-11.
6. Dodge JA, Turck D. Cystic fibrosis: nutritional consequences and management. Best Pract Res Clin Gastroenterol. 2006;20(3):531-46.
7. Alves C, Veiga S, Souza T. Dislipidemia e risco de doença cardiovascular em crianças e adolescentes com diabetes melito tipo 1. Rev Paul Pediatria. 2007; 25(1):82-9.
8. Santos V, Canadas V, Gusmão A, Almeida NC. Investigação diagnóstica das dislipidemias. In: Vilar L, editor. Endocrinologia Clínica. Rio de Janeiro: Guanabara Koogan; 2006. p.733-47.
9. Maloy MJ, Kane JP. Disorders of lipoprotein metabolism. In: Greenspan FS, Gradner DG, editors. Basic and Clinical Endocrinology. New York: MacGraw-Hill Medical; 2004. p.766-93.
10. Bricarello SG, Bricarello LP, Alves RC. Conduta diagnóstica e terapêutica nas hiperlipidemias em Pediatria. Pediatr Mod. 1999;35(12):929-47.
11. Monte O, Cisternas JR. Dislipidemias. In: Monte O, Longui CA, Calliari LE, Kochi C, editors. Endocrinologia para o pediatra. São Paulo: Ateneu; 2006. p. 573-90.
12. Volta C, Street ME, Ziveri MA, Bonelli P, Spaggiari C, Grzincich GL, et al. Thyroid function, cytokine and IGF-IGFBP interactions in cystic fibrosis patients. Horm Res. 2005;63(4):206-10.
13. Reis FJ, Damaceno N. Fibrose cística. J Pediatr (Rio J.). 1998; 74(Supl 1):S76-S94.
14. Parks EJ, Hellerstein MK. Carbohydrate-induced hypertriacylglycerolemia: historical perspective and review of biological mechanisms. Am J Clin Nutr. 2000;71(2):412-33.
15. Frase LL, Strickland AD, Kachel GW, Krejs GJ. Enhanced glucose absorption in the jejunum of patients with cystic fibrosis. Gastroenterology. 1985;88(2):478-84.
16. Slesinski MJ, Gloninger MF, Costantino JP, Orenstein DM. Lipid levels in adults with cystic fibrosis. J Am Diet Assoc. 1994;94(4):402-8. Erratum in: J Am Diet Assoc 1994;94(10):1101.
17. Burdge GC, Goodale AJ, Hill CM, Halford PJ, Lambert EJ, Postle AD, et al. Plasma lipid concentrations in children with cystic fibrosis: the value of a high-fat diet and pancreatic supplementation. Br J Nutr. 1994;71(6):959-64.
18. Kalivianakis M, Minich DM, Bijleveld CM, van Aalderen WM, Stellaard F, Laseur M, et al. Fat malabsorption in cystic fibrosis patients receiving enzyme replacement therapy is due to impaired intestinal uptake of long-chain fatty acids. Am J Clin Nutr. 1999;69(1):127-34.
19. Fagundes ED, Silva RA, Roquete ML, Penna FJ, Reis FJ, Goulart EM, et al. Validação do escore ultra-sonográfico de Williams para o diagnóstico da hepatopatia da fibrose cística. J Pediatr (Rio J). 2004;80(5):380-6.
20. Treem WR, Stanley CA. Massive hepatomegaly, steatosis, and secondary plasma carnitine deficiency in an infant with cystic fibrosis. Pediatrics. 1989;83(6):993-7.
21. Lepage G, Yesair DW, Ronco N, Champagne J, Bureau N, Chemtob S, Bérubé D, et al. Effect of an organized lipid matrix on lipid absorption and clinical outcomes in patients with cystic fibrosis. J Pediatr. 2002;141(2):178-85.
22. Levy E, Gurbindo C, Lacaille F, Paradis K, Thibault L, Seidman E. Circulating tumor necrosis factor-alpha levels and lipid abnormalities in patients with cystic fibrosis. Pediatr Res. 1993;34(2):162-6.
23. Roulet M, Frascarolo P, Rappaz I, Pilet M. Essential fatty acid deficiency in well nourished young cystic fibrosis patients. Eur J Pediatr. 1997;156(12):952-6.
24. Strandvik B. Fatty acid metabolism in cystic fibrosis. N Engl J Med. 2004;350(6):605-7.
25. Freedman SD, Blanco PG, Zaman MM, Shea JC, Ollero M, Hopper IK, et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N Engl J Med. 2004;350(6):560-9.
26. Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ. 2007;176(8):1113-20.
27. Peretti N, Marcil V, Drouin E, Levy E. Mechanisms of lipid malabsorption in cystic fibrosis: the impact of fatty acids deficiency. Nut Metab [serial on the Internet]. 2005 [cited 2005 May 3];2(1):11. Available from: http://www.nutritionandmetabolism.com/content/2/1/11.
28. Christophe AB, Warwick WJ, Holman RT. Serum fatty acid profiles in cystic fibrosis patients and their parents. Lipids. 1994;29(8):569-75.
29. Farrell PM, Mischler EH, Engle MJ, Brown DJ, Lau SM. Fatty acid abnormalities in cystic fibrosis. Pediatr Res. 1985;19(1):104-9.
30. Coste TC, Armand M, Lebacq J, Lebecque P, Wallemacq P, Leal T. An overview of monitoring and supplementation of omega 3 fatty acids in cystic fibrosis. Clin Biochem. 2007;40(8):511-20.
31. Reynders V, Loitsch S, Steinhauer C, Wagner T, Steinhilber D, Bargon J. Peroxisome proliferators-activated receptor α (PPARα) down-regulation in cystic fibrosis lymphocytes. Respir Res [serial on the Internet] 2006 [cited 2006 Oct 10]; 7:104. Available from: http://respiratory-research.com/content/7/1/104.
32. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. 2002;196(8):1025-37.
33. Durieu I, Abbas-Chorfa F, Drai J, Iwaz J, Steghens JP, Puget M, et al. Plasma fatty acids and lipid hydroperoxides increase after antibiotic therapy in cystic fibrosis. Eur Respir J. 2007;29(5):958-64.
34. Pierre M, Husson MO, Le Berre R, Desseyn JL, Galabert C, Béghin L, et al. Omega-3 polyunsaturated fatty acids improve host response in chronic Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1422-31.
35. Sarwar N, Danesh J, Eiriksdottir G, Sigurdsson G, Wareham N, Bingham S, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115(4):450-8.
36. National Cholesterol Education Program. Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Elk Grove Village: American Academy of Pediatrics, 1992.
37. Sociedade Brasileira de Cardiologia. I Diretriz de prevenção da aterosclerose na infância e na adolescência. Arq Bras Cardiol. 2005;85[Suppl 6]:S1-S36.
38. Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486-97.
39. FitzSimmons SC, Burkhart GA, Borowitz D, Grand RJ, Hammerstrom T, Durie PR, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med. 1997;336(18):1283-9.
40. Colombo C, Bennato V, Costantini D, Valmarana L, Daccò V, Zazzeron L, et al. Dietary and circulating polyunsaturated fatty acids in cystic fibrosis: are they related to clinical outcomes? J Pediatr Gastroenterol Nutr. 2006;43(5):660-5.
41. Rogers D, Prasad SA, Doull I. Exercise testing in children with cystic fibrosis. J R Soc Med. 2003;96(Suppl 43):23-9.
42. Lepage G, Paradis K, Lacaille F, Sénéchal L, Ronco N, Champagne J, et al. Ursodeoxycholic acid improves the hepatic metabolism of essential fatty acids and retinol in children with cystic fibrosis. J Pediatr. 1997;130(1):52-8.
43. Cheng K, Ashby D, Smyth R. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev. 2000;(2):CD000222.
Trabalho realizado no Serviço de Endocrinologia Pediátrica, Hospital Universitário Professor Edgard Santos, Faculdade de Medicina da Universidade Federal da Bahia - UFBA - Salvador (BA) Brasil.
1. Coordenador da Residência Médica em Endocrinologia Pediátrica. Faculdade de Medicina da Universidade Federal da Bahia - UFBA - Salvador (BA) Brasil.
2. Residente de Terceiro Ano de Endocrinologia Pediátrica. Faculdade de Medicina da Universidade Federal da Bahia - UFBA - Salvador (BA) Brasil.
Endereço para correspondência: Crésio de Aragão Dantas Alves. Rua Plínio Moscoso, 222/601, CEP 40157-190, Salvador, BA, Brasil.
Tel 55 71 9178-4055/55 71 3357-5500. E-mail: cresio.alves@uol.com.br
Apoio financeiro: Nenhum.
Recebido para publicação em 30/10/2007. Aprovado, após revisão, em 3/3/2008.












 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket